
Specialty medical genetics OMIM 106260 | ICD-10 Q82.4 DiseasesDB 33336 | |
 | ||
Hay–Wells syndrome (also known as AEC syndrome; see Naming) is one of at least 150 known types of ectodermal dysplasia.
Contents
These disorders affect tissues that arise from the ectodermal germ layer, such as skin, hair, and nails.
Genetics
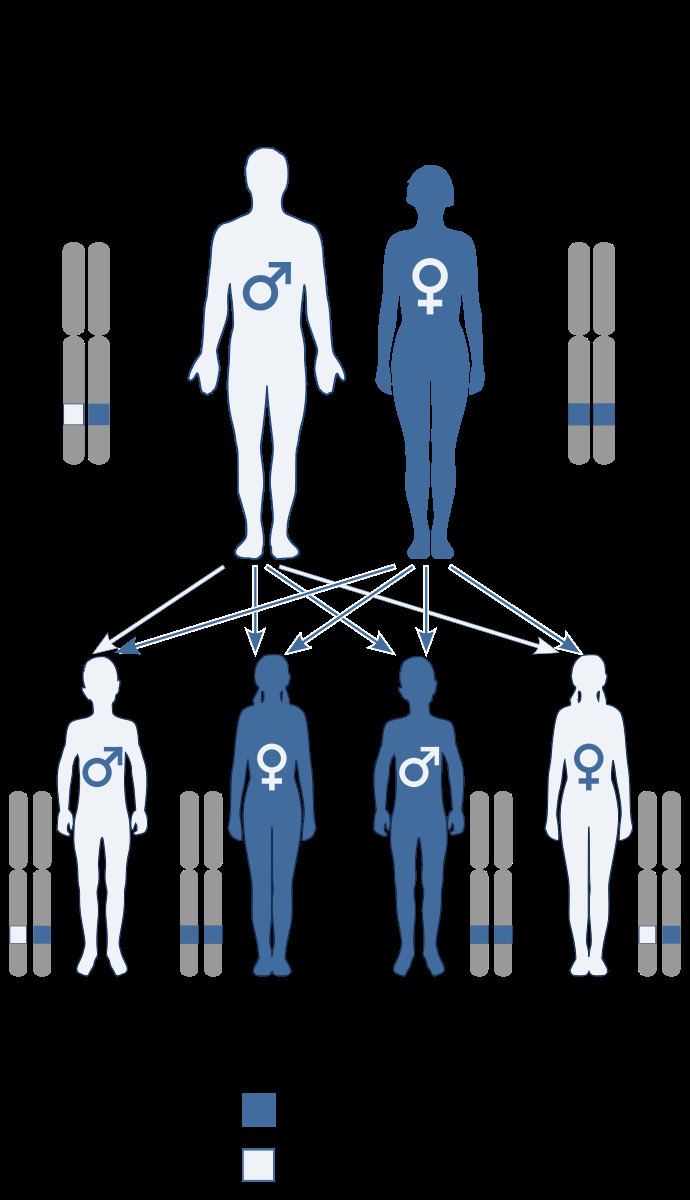
Hay–Wells syndrome is autosomal dominant, caused by a missense mutation in the Sterile alpha motif (SAM) of the TP73L (p63) gene which encodes for a protein-protein interaction domain. It is a very rare disorder.
Hay–Wells syndrome is represents an autosomal dominant pattern of inheritance. The syndrome is thought to arise from a missense mutation in a gene pivotal for the proper development of craniofacial structures and extremities, as well as skin differentiation. Specifically, mutations within the Tumor Protein 63 gene have been implicated in Hay–Wells syndrome.
Residing on the long-arm of chromosome 3, the Tumor Protein 63 (TP63) gene is critical for proper development and homeostasis of stratified epithelia. In Hay–Wells syndrome, and other ectodermal dysplasia disorders, a missense, nonsense, or insertion mutation has occurred in the TP63 gene. Currently, no deletion or duplication mutations have been detected in such disorders. Interestingly, though ectodermal dysplasia disorders result from heterozygous mutations in TP63, compromised epidermal differentiation with epidermal decay is representative of Hay-Wells patients but is hardly observed in other syndromes. In contrast, severe abnormalities characteristic of other ectodermal dysplasia disorders (i.e. limb abnormalities in EEC) are not seen in Hay-Wells patients.
Proteomics
TP63 encodes for the p63 transcription factor, which is implicated in proliferation, differentiation, apoptosis, regular cell maintenance, and cell adhesion. Specifically, p63 is expressed within early keratinocytes and the embryonic ectodermal ridge during development. Thus, p63 is believed to play a pivotal role in the development and maintenance of the epidermis. Reported mutations that have resulted in Hay–Wells syndrome have occurred within the sterile alpha motif (SAM) and the transactivation inhibitory (TI) domains of the p63-coding region. The SAM domain of p63 is thought to be imperative for protein-protein interactions, while the TI domain may play a role in the repression of other isoforms of p63. Recent work has shown that mutations within these domains lead to repression of other known transcriptional activators of epidermal differentiation. These transcription activators include: GRHL3, HOPX, PRDM1, KLF4, and ZNF750. Most notably, Hay-Wells p63 mutants irregularly repress the genes that encode for ZNF750. The down-regulation of ZNF750 has been shown to hinder the expression of the other before mentioned differentiation-activators such as HOPX, PRDM1, KLF4, and GRHL3. In contrast, recapitulating the expression of ZNF750 leads to significant rescue of normal epidermal differentiation.
Phenotype
Hay–Wells syndrome is the result of the invariant mutations of the p63 transcription factor that have been previously identified. Due to the diminished activities of p63, patients can experience a host of symptoms related to the operation of keratinocytes. In particular, the hypopigmentation observed in several Hay-Wells patients is believed to be the result of improperly developed keratinocytes not being able to properly interact with melanocytes. However, as it stands, this display of Hay–Wells syndrome has not been entirely comprehended. Most noted are the abnormal development of hair, teeth, glands, and nails.
Diagnosis
In HWS the hair is coarse and sparse, eyelashes are sparse or absent, nails may be absent or malformed, and teeth may be small and malformed. There may be fewer than normal sweat glands and they may produce little sweat, a condition known generally as hypohidrosis. Chronic inflammatory dermatitis of the scalp is a common symptom.
Two features differentiate HWS from other ectodermal displasias. First, the syndrome is associated with cleft palate, and, less often, cleft lip. Second, the edges of the upper and lower eyelid grow bands of fibrous tissue, often causing them to be fused together. This condition in the eyelids is called ankyloblepharon filiforme adnatum.
Naming
Hay–Wells syndrome is also known as AEC syndrome; this is short for "ankyloblepharon–ectodermal dysplasia–clefting syndrome", "ankyloblepharon filiforme adnatum–ectodermal dysplasia–cleft palate syndrome", "ankyloblepharon–ectodermal defects–cleft lip/palate (AEC) syndrome", "ankyloblepharon–ectodermal defect–cleft lip and/or palate syndrome", or "ankyloblepharon ectodermal dysplasia and clefting". Hay–Wells syndrome, or Ankyloblepharon-Ectodermal Dysplasia-Clefting (AEC) syndrome, is one of over one-hundred forms of ectodermal dysplasia; a collection of inherited diseases that cause atypical development of nails, glands, teeth, and hair. Males and females are equally affected by Hay–Wells syndrome. No demographic has been shown to be especially susceptible to the syndrome. In the United States, Hay-Wells like syndromes occur in only one in 100,000 births. Symptoms are apparent at birth, or become apparent when atypical development of teeth occurs. Major symptoms of Hay–Wells syndrome include: sparse hair and eyelashes, missing teeth, cleft palate, cleft lip with fusing of the upper and lower eyelids, and deformed nails. Therefore, a diagnosis of Hay–Wells syndrome is largely based upon the physical clinical presentation of the patient.