
Specialty Haematology ICD-9-CM 286.1 DiseasesDB 5561 | ICD-10 D67 OMIM 306900 MedlinePlus 000539 | |
 | ||
Haemophilia B (or hemophilia B) is a blood clotting disorder caused by a mutation of the factor IX gene, leading to a deficiency of factor IX. It is the second-most common form of haemophilia, rarer than haemophilia A. Haemophilia B was first recognized as a different kind of haemophilia in 1952. It is sometimes called Christmas disease, named after Stephen Christmas, the first patient described with this disease. In addition, the first report of its identification was published in the Christmas edition of the British Medical Journal.
Contents
Signs/symptoms
The presentation of hemophilia B is consistent with easy bruising, urinary tract bleed and nosebleeds. Additionally, the affected individual may experience bleeding into their joints.
Genetics
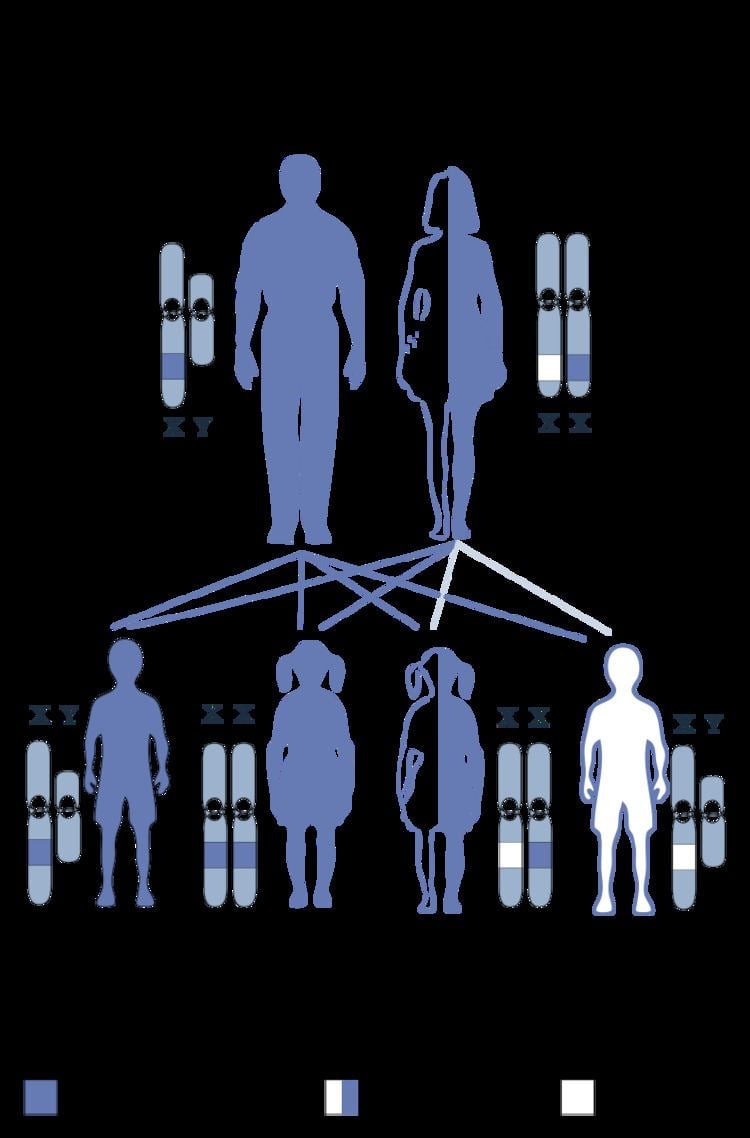
The factor IX gene is located on the X chromosome (Xq27.1-q27.2). It is an X-linked recessive trait, which explains why, as in haemophilia A, usually only males are affected.
In 1990, George Brownlee and Merlin Crossley showed that two sets of genetic mutations were preventing two key proteins from attaching to the DNA of people with a rare and unusual form of haemophilia B – haemophilia B Leyden – where sufferers experience episodes of excessive bleeding in childhood but have few bleeding problems after puberty. This lack of protein attachment to the DNA was thereby turning off the gene that produces clotting factor IX, which prevents excessive bleeding.
Pathophysiology
In terms of mechanism, factor IX deficiency leads to an increased propensity for haemorrhage. This is in response to mild trauma or even spontaneously, such as in joints (haemarthrosis) or muscles.Factor IX deficiency can cause interference of the coagulation cascade, thereby causing spontaneous hemorrhage when there is trauma. Factor IX when activated activates factor X which helps fibrinogen to fibrin conversion.
Factor IX becomes active eventually in coagulation, by cofactor factor VIII (specifically IXa) Platelets provide a binding site for both cofactors. This complex (in the coagulation pathway) will eventually activate factor X.
Diagnosis
The diagnosis for hemophilia B can be done via the following tests/methods:
Differential diagnosis
The differential diagnosis for this inherited condition is the following: hemophilia A, factor XI deficiency, von Willebrand disease, fibrinogen disorders and Bernard-Soulier syndrome
Treatment
Treatment is by intravenous infusion of factor IX, which has a longer half life than factor VIII and as such factor IX can be transfused less frequently. Blood transfusions may be needed, NSAIDS should be discontinued once the individual has been diagnosed with the condition.Any surgical procedure should be done in concert with tranexamic acid.
Prognosis
Two Dutch studies have followed hemophilia patients for a number of years. Both studies found that viral infections were common in hemophiliacs due to the frequent blood transfusions which put them at risk of getting blood borne infections such as HIV and hepatitis C. In the latest study which followed patients from 1992 to 2001, the male life expectancy was 59 years. If cases with known viral infections were excluded, the life expectancy was 72, close to that of the general population. 26% of the cases died from AIDS and 22% from hepatitis C.
History
In the 1950s and 1960s, with newfound technology and gradual advances in medicine, pharmaceutical scientists found a way to take the factor IX from fresh frozen plasma (FFP) and give it to those with haemophilia B. Though they found a way to treat the disease, the FFP contained only a small amount of factor IX, requiring large amounts of FFP to treat an actual bleeding episode, which resulted in the person requiring hospitalization. By the mid-1960s scientists found a way to get a larger amount of factor IX from FFP. By the late 1960s, pharmaceutical scientists found methods to separate the factor IX from plasma, which allows for neatly packaged bottles of factor IX concentrates. With the rise of factor IX concentrates it became easier for people to get treatment at home. Although these advances in medicine had a significant positive impact on the treatment of haemophilia, there were many complications that came with it. By the early 1980s, scientists discovered that the medicines they had created were transferring blood-borne viruses, such as hepatitis, and HIV, the virus that causes AIDS. With the rise of these deadly viruses, scientists had to find improved methods for screening the blood products they received from donors.In 1982, scientists made a breakthrough in medicine and were able to clone factor IX gene. With this new development it decreased the risk of the many viruses. Although the new factor was created, it wasn't available for haemophilia B patients till 1997.
Society
A study published in 2009 identified the blood disease affecting the royal families of Great Britain, Germany, Russia and Spain as haemophilia B on the basis of genetic markers.