
Specialty endocrinology ICD-9-CM 271.0 DiseasesDB 5303 | ICD-10 E74.0 OMIM 232500 263570 607839 eMedicine med/910 ped/97 | |
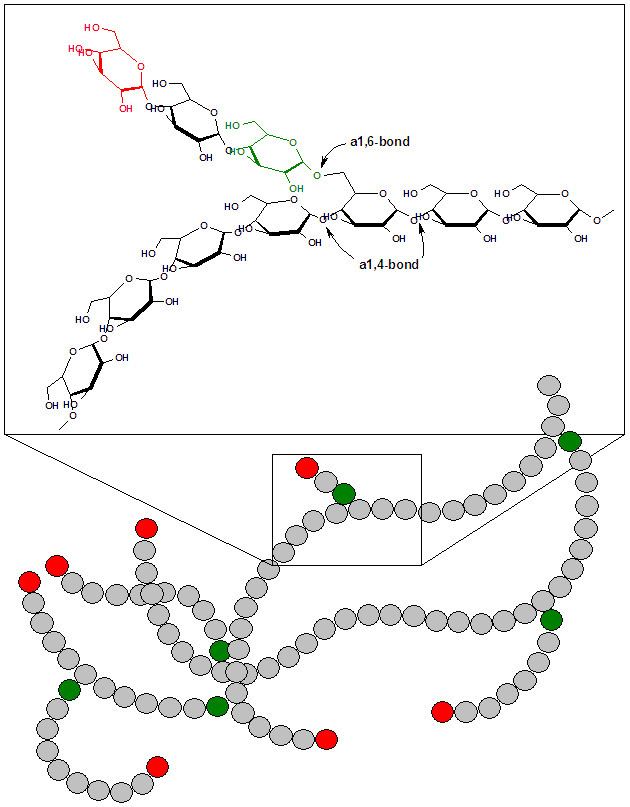 | ||
Glycogen storage disease type IV is a form of glycogen storage disease, which is caused by an inborn error of metabolism. It is the result of a mutation in the GBE1 gene, which causes a defect in the glycogen branching enzyme. Therefore, glycogen is not made properly and abnormal glycogen molecules accumulate in cells; most severely in cardiac and muscle cells. The severity of this disease varies on the amount of enzyme produced. Glycogen Storage Disease Type IV is autosomal recessive, which means each parent has a mutant copy of the gene but show no symptoms of the disease. It affects 1 in 800,000 individuals worldwide, with 3% of all Glycogen Storage Diseases being type IV.
Contents
Names
It is also known as:
The eponym "Andersen's disease" is sometimes used, for Dorothy Hansine Andersen.
Mutations in GBE1 can also cause a milder disease in adults called adult polyglucosan body disease.
Human pathology
It is a result of the absence of the glycogen branching enzyme, which is critical in the production of glycogen. This leads to very long unbranched glucose chains being stored in glycogen. The long unbranched molecules have a low solubility which leads to glycogen precipitation in the liver. These deposits subsequently build up in the body tissue, especially the heart and liver. The inability to breakdown glycogen in muscle cells causes muscle weakness. The probable end result is cirrhosis and death within 5 years. In adults, the activity of the enzyme is higher and symptoms do not appear until later in life.
Fatal perinatal neuromuscular type
Congenital muscular type
Progressive hepatic type
Non-progressive hepatic type
Childhood neuromuscular type
In animals
In horses: it has been reported in American Quarter Horses and related breeds.
In cats: the disease has been reported in the Norwegian Forest Cat, where it causes skeletal muscle, heart, and CNS degeneration in animals greater than 5 months old. It has not been associated with cirrhosis or liver failure.