
EC number 2.4.1.18 ExPASy NiceZyme view | CAS number 9001-97-2 | |
 | ||
Glycogen branching enzyme is an enzyme that adds branches to the growing glycogen molecule during the synthesis of glycogen, a storage form of glucose. More specifically, during glycogen synthesis, a glucose 1-phosphate molecule reacts with uridine triphosphate (UTP) to become UDP-glucose, an activated form of glucose. The activated glucosyl unit of UDP-glucose is then transferred to the hydroxyl group at the C-4 of a terminal residue of glycogen to form an α-1,4-glycosidic linkage, a reaction catalyzed by glycogen synthase. Importantly, glycogen synthase can only catalyze the synthesis of α-1,4-glycosidic linkages. Since glycogen is a readily mobilized storage form of glucose, the extended glycogen polymer is branched by glycogen branching enzyme to provide glycogen breakdown enzymes, such as glycogen phosphorylase, with a large number of terminal residues for rapid degradation. Branching also importantly increases the solubility and decreases the osmotic strength of glycogen.
Contents
Nomenclature
This enzyme belongs to the family of transferases, to be specific, those glycosyltransferases that transfer hexoses (hexosyltransferases). The systematic name of this enzyme class is 1,4-alpha-D-glucan:1,4-alpha-D-glucan 6-alpha-D-(1,4-alpha-D-glucano)-transferase. Other names in common use include branching enzyme, amylo-(1,4→1,6)-transglycosylase, Q-enzyme, alpha-glucan-branching glycosyltransferase, amylose isomerase, enzymatic branching factor, branching glycosyltransferase, enzyme Q, glucosan transglycosylase, 1,4-alpha-glucan branching enzyme, plant branching enzyme, alpha-1,4-glucan:alpha-1,4-glucan-6-glycosyltransferase, and starch branching enzyme. This enzyme participates in starch and sucrose metabolism.
Gene
GBE is encoded by the GBE1 gene.
Through southern blot analysis of DNA derived from human/rodent somatic cell hybrids, GBE1 has been identified as an autosomal gene located on the short arm of chromosome 3 at position 12.3. The human GBE gene was also isolated by a function complementation of the Saccharomyces cerevisiae GBE deficiency. From the isolated cDNA, the length of the gene was found to be approximately 3 kb. Additionally, the coding sequence was found to comprise 2,106 base pairs and encode a 702-amino acid long GBE. The molecular mass of human GBE was calculated to be 80,438 Da.
Structure
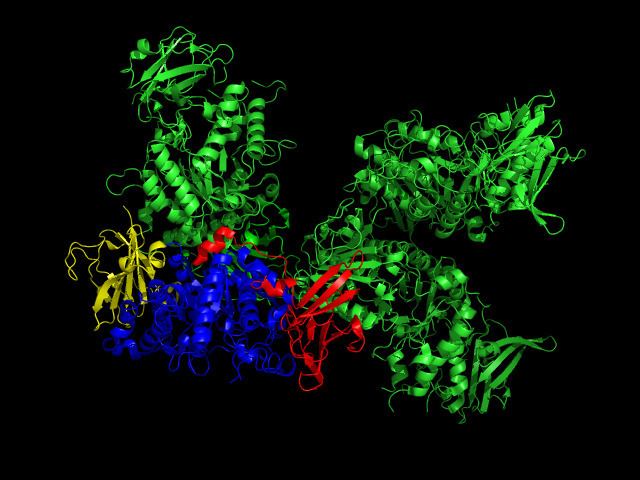
Glycogen branching enzyme belongs to the α-amylase family of enzymes, which include α-amylases, pullulanas/isoamylase, cyclodextrin glucanotransferase (CGT), and branching enzyme. Shown by x-ray crystallography, glycogen branching enzyme has four marginally asymmetric units each that are organized into three domains: an amino-terminal domain, involved in determining the length of the chain transfer, a carboxyl-terminal domain, involved in substrate preference and catalytic capacity, and a central (α/β) barrel catalytic domain. The amino-terminal domain consists of 128 residues arranged in seven β-strands, the carboxyl-terminal domain with 116 residues also organized in seven β-strands, and the (α/β) barrel domain with 372 residues. While the central (α/β) barrel domain is common in members of the α-amylase family, numerous variations exist between the various barrel domains. Additionally, there are striking differences between the loops connecting elements of the secondary structure among these various α-amylase members, especially around the active site. In comparison to the other family members, glycogen binding enzyme has shorter loops, which result in a more open cavity, favorable to the binding of a bulkier substrate such as branched sugar. Through primary structure analysis and the x-ray crystallographic structures of the members of the α-amylase family, seven residue were conserved, Asp335, His340, Arg403, Asp 405, Glu458, His525, and Asp526 (E coli. numbering). These residues are implicated in catalysis and substrate binding.
Glycogen binding enzymes in other organisms have also been crystallized and structurally determined, demonstrating both similarity and variation to GBE found in Escherichia coli.
Function
In glycogen, every 10 to 14 glucose units, a side branch with an additional chain of glucose units occurs. The side chain attaches at carbon atom 6 of a glucose unit, an α-1,6-glycosidic bond. This connection is catalyzed by a branching enzyme, generally given the name α-glucan branching enzyme. A branching enzyme attaches a string of seven glucose units (with some minor variation to this number) to the carbon at the C-6 position on the glucose unit, forming the α-1,6-glycosidic bond. The specific nature of this enzyme means that this chain of 7 carbons is usually attached to a glucose molecule that is in position three from the non-reducing end of another chain. Because the enzyme works with such specificity regarding the number of glucose units transferred and the position to which they are transferred, the enzyme creates the very characteristic, highly branched glycogen molecule.
Clinical significance
Mutations in this gene are associated with glycogen storage disease type IV (also known as Andersen's disease) in newborns and with adult polyglucosan body disease.
Approximately 40 mutations in the GBE1 gene, most resulting in a point mutation in the glycogen branching enzyme, have led to the early childhood disorder, glycogen storage disease type IV (GSD IV). This disease is characterized by a severe depletion or complete absence of GBE, resulting in the accumulation of abnormally structured glycogen, known as polyglucosan bodies. Glycogen buildup leads to increased osmotic pressure resulting in cellular swelling and death. The tissues most affected by this disease are the liver, heart, and neuromuscular system, areas with the greatest levels of glycogen accumulation. Abnormal glycogen buildup in the liver interferes with liver functioning and can result in an enlarged liver and liver disease. In muscles, the inability of cells to efficiently breakdown glycogen due to the severe reduction or absence of branching can lead to muscle weakness and atrophy. At least three mutations in the GBE1 gene have been found to cause another disease called adult polyglucosan body disease (APBD). While in GSD IV GBE activity is undetectable or minimally detectable, APBD is characterized by reduced or even normal GBE activity. In this disease, abnormal glycogen can build up in neurons leading to a spectrum of problems. Specifically, some disease characteristics are gait difficulties from mixed upper and lower motor neuron involvement sensory loss in lower extremities, and neurogenic bladder, a problem in which a person lacks bladder control due to a brain, spinal cord, or nerve condition.
Model organisms
Model organisms have been used in the study of GBE1 function. A conditional knockout mouse line, called Gbe1tm1a(KOMP)Wtsi was generated as part of the International Knockout Mouse Consortium program — a high-throughput mutagenesis project to generate and distribute animal models of disease to interested scientists.
Male and female animals underwent a standardized phenotypic screen to determine the effects of deletion. Twenty six tests were carried out on mutant mice and two significant abnormalities were observed. No homozygous mutant embryos were identified during gestation, and therefore none survived until weaning. The remaining tests were carried out on heterozygous mutant adult mice; no additional significant abnormalities were observed in these animals.