
 | ||
Phosphodiesterases (PDEs) are a superfamily of enzymes. This superfamily is further classified into 11 families, PDE1 - PDE11, on the basis of regulatory properties, amino acid sequences, substrate specificities, pharmacological properties and tissue distribution. Their function is to degrade intracellular second messengers such as cyclic adenine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) which leads to several biological processes like effect on intracellular calcium level by the Ca2+ pathway.
Contents
- Discovery
- Penile erection
- Erectile dysfunction
- The phosphodiesterase 5 enzyme
- PDE5 Inhibitors
- Side effects
- Structureactivity relationship SAR
- Other research
- References
Phosphodiesterase 5 (PDE5) is widely expressed in several tissues in the body for example brain, lung, kidney, urinary bladder, smooth muscle and platelets. It is possible to prevent cGMP hydrolysis by inhibiting PDE5 and therefore treat diseases associated with low cGMP levels, because of this, PDE5 is an ideal target for the development of inhibitors.
The three major PDE5 inhibitors are sildenafil, tadalafil and vardenafil.
Discovery
PDE5 is an enzyme that was first purified in 1980 from a rats lung. PDE5 converts intracellular cGMP to the nucleotide GMP. Many tissues contain PDE5, such as lungs, kidneys, brain, platelets, liver, prostate, urethra, bladder and smooth muscles. Because of the localization of PDE5 in the smooth muscle tissue, inhibitors were developed for the treatment of erectile dysfunction along with pulmonary hypertension.
Sildenafil was initially introduced for clinical trial in 1989. It was the result of extensive research on chemical agents targeting PDE5 that could be effective in treatment of coronary heart disease. Sildenafil did not prove effective for coronary heart disease but an interesting side effect was discovered, a penile erection. That side effect soon became the main field of investigation. The inhibitor is highly selective for the PDE5 family.
Sildenafil is a prototype of PDE5 inhibitors that Pfizer launched as Viagra. It was approved by the Food and Drug Administration (FDA) in 1998 as the first oral medicine for erectile dysfunction. Later, in the year 2005, it was approved for the treatment of pulmonary arterial hypertension. Vardenafil and tadalafil were discovered in 1990. These drugs came out of research programs focusing on finding PDE5 inhibitors for the treatment of cardiovascular diseases and erectile dysfunction.The two PDE5 inhibitors soon became treatments for these conditions.
Tadalafil is the most versatile inhibitor and has the longest half-life, 17,5 hours. This allows for a longer therapeutic window and is therefore often a more convenient drug than others with a shorter therapeutic window. Tadalafil is more bioavailable (80%) than sildenafil (40%) and vardenafil (15%) but it has a slow absorption, or about 2 hours compared to 50 minutes of sildenfil. Vardenafil is most known for its potency.
Because of severe adverse effects and patients dissatisfaction with current therapy choices other inhibitors have recently been approved for clinical use. These inhibitors are udenfil, avanafil lodenafil and mirodenafil.
Penile erection
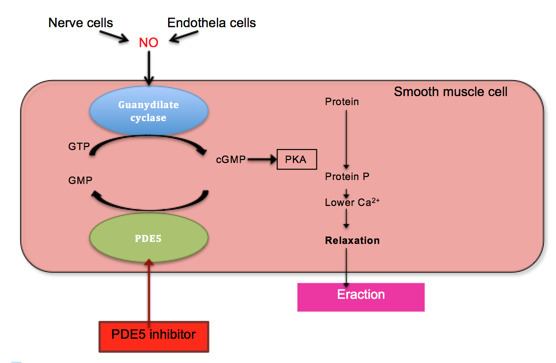
Penile erection is a hemodynamic event in the smooth muscle of corpus cavernous. PDE5 is the main cGMP hydrolysing enzyme found in penile corpus cavernous. Erection is triggered by release of the neurotransmitter nitric oxide (NO) from non-adrenergic and non-cholinergic neurons from nerve ending in the penis as well as from endothelial cells. NO activates soluble guanylyl cyclase in smooth muscle cells in the penis which results in increased production of 3'-5'-cyclic guanosine monophosphate from guanosine-5'-triphosphate (GTP). Cyclic GMP binds to the cGMP-dependent protein kinase (PKG1) which phosphorylates several proteins that results in decreased intracellular calcium. Lower intracellular calcium leads to smooth muscle relaxation and ultimately penile erection. This pathway is demonstrated in figure 1.
Erectile dysfunction
PDE5 degrades cGMP and therefore inhibits erection. As demonstrated in figure 1, inhibition of PDE5 reduces degradation of cGMP and leads to penile erection. Because of this action PDE5 inhibitors have been developed for the treatment of penile erectile dysfunction.
The phosphodiesterase 5 enzyme
The PDE5 enzyme has a molecular mass of 200 kDa and its active state is a homodimer. PDE5 consists of monomers and each contains two major functional domains: the regulatory domain (R domain) which is located in the N-terminal portion of the protein and the catalytic domain (C domain) located in the more C-terminal portion of the protein.
The R domain contains specific allosteric cGMP binding site that controls the enzymes function. This specific binding site consists of subdomain GAF (cGMP-specific cGMP-stimulated PDE, adenylate cyclase, and FhlA) which is located in the N-terminal section of the specific proteins. The allosteric binding site GAF consists of GAFa and GAFb where GAFa has a higher binding affinity. The importance and functional role of the two homologous binding sites are unknown.
Conformational change occurs when cGMP binds to the allosteric site that exposes serine and permits phosphorylation. The results for the phosphorylation of serine leads to increased cGMP hydrolysis at the catalytic domain. The affinity of the catalytic domain for cGMP increases and further increases the PDE5 catalytic domain activity. Through the C domain, intracellular cGMP is degraded rapidly by PDE5 which minimizes the activity of cGMP on its PKG1 substrate by cleaving the cyclic phosphate part of cGMP to GMP. GMP is an inactive molecule with no second messenger activity.
Phosphorylation of a single serine by PKG1 and the allosteric cGMP binding site activates the PDE5 catalytic activity and the result is a negative feedback regulation of cGMP/NO/PKG1 signalling. cGMP therefore interacts with both allosteric and catalytic domain of the PDE5 enzyme and PDE5 inhibitors compete with cGMP for binding at the catalytic domain resulting in higher cGMP levels. PDE5 domains are demonstrated in figure 2.
PDE5 Inhibitors
The PDE5 inhibitors sildenafil, vardenafil and tadalafil are competitive and reversible inhibitors of cGMP hydrolysis by the catalytic side of PDE5. The structures of vardenafil and sildenafil are similar, they both contain similar structured purine ring of cGMP that contributes their features to act as a competitive inhibitor of PDE5. The difference of the molecular structures is the reason for interaction with the catalytic site of PDE5 and improves the affinity of these compounds compared with cGMP selectivity.
Pharmacophore
The pharmacophore model of PDE5 usually consists of one hydrogen bond acceptor, one hydrophobic aliphatic carbon chain and two aromatic rings. Small hydrophobic pocket and H-loop of PDE5 enzyme are important for binding affinity of PDE5 inhibitors. As well as positional and conformational changes are observed upon inhibitor binding in many cases.
The active site of PDE5 is located at a helical bundle domain at the center of C domain (catalytic domain). The substrate pocket is composed of four subsites: M site (metal-binding site), Q pocket (core pocket), H pocket (hydrophobic pocket) and L region (lid region) as demonstrated in figure 3. The Q pocket accommodates the pyrazolopyrimidinone group of sildenafil. That suggest that other chemicals similar to guanidine groups of cGMP can also bind at this region. The amino acids residues, Gln817, Phe820, Val782 and Tyr612, are lined in the Q pocket, they are highly conserved in all PDEs. The amide moiety of the pyrazolopyrimidinone group forms a bidentate hydrogen bond with the ɣ-amide group of Gln817. 3D structure of sildenafil is demonstrated in figure 4.
Side effects
No serious side effects have occurred in clinical trials although serious adverse effects have been recognized. For those who are taking nitrates parallel PDE5 inhibitors systemic hypotension may occur and therefore patients should not take nitrates with PDE5 inhibitors. Hearing inpairment is one risk factor for those who are using PDE5 inhibitors and it has been reported for all three available drugs on the market. This problem may be due to high level effect cGMP on cochlear hair cells. It has been reported that PDE5 inhibitors (sildenafil & vardenafil) cause moderate-severe visual disturbances likely due to PDE6 inhibition, but recent large trial and case review found no increase in ocular risk in patient taking sildenafil.
These side effects can be attributed to the limited selectivity of PDE5 inhibitors against other PDE isozyme such as PDE1 and PDE6. That is why it is important to improve selectivity of PDE5 inhibitors that potentially would lead to fewer side effects. Vardenafil and Tadalafil have demonstrated reduced adverse effects probably due to improved selectivity for PDE5.
Several reports are about approaches to improve PDE5 inhibitors, where as chemical groups have been switched out to increase potency and selectivity, which should potentially lead to drugs with fewer side effects.
Structure–activity relationship (SAR)
Sildenafil, the first PDE5 inhibitor, was discovered through rational drug design programme. The compound was potent and selective over PDE5 but was lacking preferable pharmacological properties.
Structure-activity relationship (SAR) is demonstrated in figure 5, figure 6 and figure 7. Figure 5 demonstrates the three main groups of sildenafil, R1, R2 and R3. R1 is the pyrazolopyrimidinone ring, R2 the ethoxyphenyl ring and R3 is the methylpiperazine ring. R1 group is responsible for the binding of the drug to its active binding site of PDE5.
Solubility is one of the pharmacological properties that was increased. A group was substituted for the hydrogen atom as demonstrated in figure 6. The sulfonamide group was chosen to lower lipophilicity and increase solubility as seen in figure 7.
Solubility was further increased by placing a methyl group at R positions as demonstrated in figure 7. Other phosphodiesterase-5 inhibitors were developed from the structure in figure 7.
Other research
Although PDE5 inhibitors main use has been for erectile dysfunction there has been a great interest in PDE5 inhibitors as a promising new therapeutic agents for treatment of other diseases, such as Alzheimer's disease. Elevation of cGMP levels through inhibition of PDE5 provides a way of improving memory and learning. PDE5 has also been considered as a potential therapeutic agent for parasitic disease such as African sleeping sickness. Strategic changes were made to the structure of sildenafil so the molecule could project into a parasite-specific pocket (the p-pocket). Similar approach has be used to design therapeutic agents Plasmodium falciparum.