
 | ||
A biosimilar (also known as follow-on biologic or subsequent entry biologic) is a biologic medical product which is almost an identical copy of an original product that is manufactured by a different company. Biosimilars are officially approved versions of original "innovator" products, and can be manufactured when the original product's patent expires. Reference to the innovator product is an integral component of the approval.
Contents
- Approval processes
- Background
- European approvals of biosimilars
- BPCI Act
- Data exclusivity
- Nomenclature
- Market implications
- References
Unlike with generic drugs of the more common small-molecule type, biologics generally exhibit high molecular complexity, and may be quite sensitive to changes in manufacturing processes. Follow-on manufacturers do not have access to the originator's molecular clone and original cell bank, nor to the exact fermentation and purification process, nor to the active drug substance, although they do have access to the commercialized innovator product. Overall, it is harder to ascertain fungibility between generics and innovators among biologics than it is among totally synthesized and semisynthesized drugs, which is why the name "biosimilar" was coined to differentiate these drugs from small-molecule generics. A simple analogy is that it is harder to say that two wines are "sufficiently interchangeable", because of differences in yeast strain, weather, grape harvest, or terroir, than it is to say that two soda pops are "sufficiently interchangeable" because they contain the same flavoring powder and salts.
Drug related authorities such as European Medicines Agency (EMA), Food and Drug Administration (FDA), and Health Canada hold their own guidance on requirements for demonstration of the similar nature of two biological products in terms of safety and efficacy. According to them, analytical studies that demonstrate that the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components, animal studies (including the assessment of toxicity), and a clinical study or studies (including the assessment of immunogenicity and pharmacokinetics or pharmacodynamics) are sufficient to demonstrate safety, purity, and potency in one or more appropriate conditions of use for which the reference product is licensed and intended to be used and for which licensure is sought for the biological product.
In case of a monoclonal antibody-containing medicinal product, such as Remsima, extensive physicochemical and biological characterization for it and its reference product Remicade was conducted in order to demonstrate their highly similar properties. Consequently, EMA has granted a marketing authorisation for only a few biosimilars since 2006 including a monoclonal antibody which is recently approved. Meanwhile, on March 6, 2015, the FDA approved the United States's first biosimilar product, the biosimilar of filgrastim called filgrastim-sndz (trade name Zarxio) by Sandoz.
Approval processes
The European regulatory authorities led with a specially adapted approval procedure to authorize subsequent versions of previously approved biologics, termed "similar biological medicinal products", or biosimilars. This procedure is based on a thorough demonstration of "comparability" of the "similar" product to an existing approved product. In the United States, the Food and Drug Administration (FDA) held that new legislation was required to enable them to approve biosimilars to those biologics originally approved through the PHS Act pathway. Additional Congressional hearings have been held. On March 17, 2009, the Pathway for Biosimilars Act was introduced in the House. See the Library of Congress website and search H.R. 1548 in 111th Congress Session. Since 2004 the FDA has held a series of public meetings on biosimilars.
The FDA gained the authority to approve biosimilars (including interchangeables that are substitutable with their reference product) as part of the Patient Protection and Affordable Care Act signed by President Obama on March 23, 2010.
The FDA has previously approved biologic products using comparability, for example, Omnitrope in May 2006, but this like Enoxaparin was also to a reference product, Genotropin, originally approved as a biologic drug under the FD&C Act.
On March 6, 2015, Zarxio obtained the first approval of FDA. Sandoz’s Zarxio is biosimilar to Amgen’s Neupogen (filgrastim), which was originally licensed in 1991. This is the first product to be passed under the Biologics Price Competition and Innovation Act of 2009 (BPCI Act), which was passed as part of the Affordable Healthcare Act. But Zarxio was approved as a biosimilar, not as an interchangeable product, the FDA notes. And under the BPCI Act, only a biologic that has been approved as an “interchangeable” may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product. The FDA said its approval of Zarxio is based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data that demonstrates Zarxio is biosimilar to Neupogen.
Background
Cloning of human genetic material and development of in vitro biological production systems has allowed the production of virtually any recombinant DNA based biological substance for eventual development of a drug. Monoclonal antibody technology combined with recombinant DNA technology has paved the way for tailor-made and targeted medicines. Gene- and cell-based therapies are emerging as new approaches.
Recombinant therapeutic proteins are of a complex nature (composed of a long chain of amino acids, modified amino acids, derivatized by sugar moieties, folded by complex mechanisms). These proteins are made in living cells (bacteria, yeast, animal or human cell lines). The ultimate characteristics of a drug containing a recombinant therapeutic protein are to a large part determined by the process through which they are produced: choice of the cell type, development of the genetically modified cell for production, production process, purification process, formulation of the therapeutic protein into a drug.
After the expiry of the patent of approved recombinant drugs (e.g., insulin, human growth hormone, interferons, erythropoietin, monoclonal antibodies and more) any other biotech company can develop and market these biologics (thus called biosimilars). Every biological (or biopharmaceutical products) displays a certain degree of variability, even between different batches of the same product, which is due to the inherent variability of the biological expression system and the manufacturing process. Any kind of reference product has undergone numerous changes in its manufacturing processes, and such changes in the manufacturing process (ranging from a change in the supplier of cell culture media to new purification methods or new manufacturing sites) was substantiated with appropriate data and was approved by the EMA. In contrast, it is mandatory for biosimilars to take a both non-clinical and clinical test that the most sensitive clinical models are asked to show to enable detection of differences between the two products in terms of human pharmacokinetics (PK) and pharmacodynamics (PD), efficacy, safety, and immunogenicity.
The current concept of development of biosimilar mAbs follows the principle that an extensive state of the art physicochemical, analytical and functional comparison of the molecules is complemented by comparative non-clinical and clinical data that establish equivalent efficacy and safety in a clinical "model" indication that is most sensitive to detect any minor differences (if these exist) between biosimilar and its reference mAb also at the clinical level.
The European Medicines Agency (EMA) has recognized this fact, which has resulted in the establishment of the term "biosimilar" in recognition that, whilst biosimilar products are similar to the original product, they are not exactly the same. Every biological displays a certain degree of variability. However, provided that structure and function(s), pharmacokinetic profiles and pharmacodynamic effect(s) and/or efficacy can be shown to be comparable for the biosimilar and the reference product, those adverse drug reactions which are related to exaggerated pharmacological effects can also be expected at similar frequencies.
Originally the complexity of biological molecules led to requests for substantial efficacy and safety data for a biosimilar approval. This has been progressively replaced with a greater dependence on assays, from quality through to clinical, that show assay sensitivity sufficient to detect any significant difference in dose. However, the safe application of biologics depends on an informed and appropriate use by healthcare professionals and patients. Introduction of biosimilars also requires a specifically designed pharmacovigilance plan. It is difficult and costly to recreate biologics because the complex proteins are derived from living organisms that are genetically modified. In contrast, small molecule drugs made up of a chemically based compound can be easily replicated and are considerably less expensive to reproduce. In order to be released to the public, biosimilars must be shown to be as close to identical to the parent innovator biologic product based on data compiled through clinical, animal, analytical studies and conformational status.
Generally, once a drug is released in the market by FDA, it has to be re-evaluated for its safety and efficacy once every six months for the first and second years. Afterward, re-evaluations are conducted yearly, and the result of the assessment should be reported to authorities such as FDA. Biosimilars are required to undergo pharmacovigilance (PVG) regulations as its reference product. Thus biosimilars approved by EMEA (European Medicines Agency) are required to submit a risk management plan (RMP) along with the marketing application and have to provide regular safety update reports after the product is in the market. The RMP includes the safety profile of the drug and proposes the prospective pharmacovigilance studies.
Several PK studies, such as studies conducted by Committee for Medicinal Products for Human Use (CHMP), have been conducted under various ranges of conditions; Antibodies from an originator’s product versus antibodies from an biosimilar; combination therapy and monotherapy; various diseases, etc. on the purpose to verify comparability in pharmacokinetics of the biosimilar with the reference medicinal product in a sufficiently sensitive and homogeneous population. Importantly, provided that structure and function(s), pharmacokinetic profiles and pharmacodynamic effect(s) and/or efficacy can be shown to be comparable for the biosimilar and the reference product, those adverse drug reactions which are related to exaggerated pharmacological effects can also be expected at similar frequencies.
European approvals of biosimilars
It has approved (in Jan 2016) Benepali a biosimilar to Enbrel (etanercept).
BPCI Act
The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) was originally sponsored and introduced on June 26, 2007, by Senator Edward Kennedy (D-MA). It was formally passed under the Patient Protection and Affordable Care Act (PPAC Act), signed into law by President Barack Obama on March 23, 2010. The BPCI Act was an amendment to the Public Health Service Act (PHS Act) to create an abbreviated approval pathway for biological products that are demonstrated to be highly similar (biosimilar) to a Food and Drug Administration (FDA) approved biological product. The BPCI Act is similar, conceptually, to the Drug Price Competition and Patent Term Restoration Act of 1984 (also referred to as the "Hatch-Waxman Act") which created biological drug approval through the Federal Food, Drug, and Cosmetic Act (FFD&C Act). The BPCI Act aligns with the FDA's longstanding policy of permitting appropriate reliance on what is already known about a drug, thereby saving time and resources and avoiding unnecessary duplication of human or animal testing. The FDA has released a total of four draft guidelines related to biosimilar or follow-on biologics development. Upon the release of the first three guidance documents the FDA held a public hearing on May 11, 2012.
Data exclusivity
Data exclusivity is an important piece of the amendment in the Patient Protection and Affordable Care Act for biosimilars. It is the period of time between FDA approval and an abbreviated filing for a biosimilar on the original producer's data. Data exclusivity is designed to preserve innovation and recognize the long, costly, and risky process involved while the innovator waits to gain FDA approval. The time allowed for data exclusivity is critical for the future of biologics. A number of provisions for data exclusivity in recent legislative proposals ranged up to 14 years, however, the passing of the PPAC Act guarantees a 12-year time period from the time of FDA approval. This is supposed to compensate for perceived shortcomings in patent protection for biologics. Data exclusivity extends from the date of product approval, and this protection period runs concurrently with any remaining patent term protection for the biologic. That is to say, data exclusivity provides additional protection to the innovator when the remaining patent length is shorter than the data exclusivity period at the time of approval (which can occur due to lengthy pre-clinical and clinical research required to obtain FDA approval), or to the extent that the patent term is circumvented by a biosimilar prior to its expiry.
Nomenclature
The World Health Organization (WHO) and the FDA have been working for years on the nonproprietary naming of biosimilars. In August 2015, the FDA published a draft guideline on the topic. In brief, the guideline calls for the assignment of a four character alphabetic suffix to the nonproprietary name of the original product to distinguish between innovator drugs and their biosimilars. The WHO INN system calls this suffix a biologic modifier.
Market implications
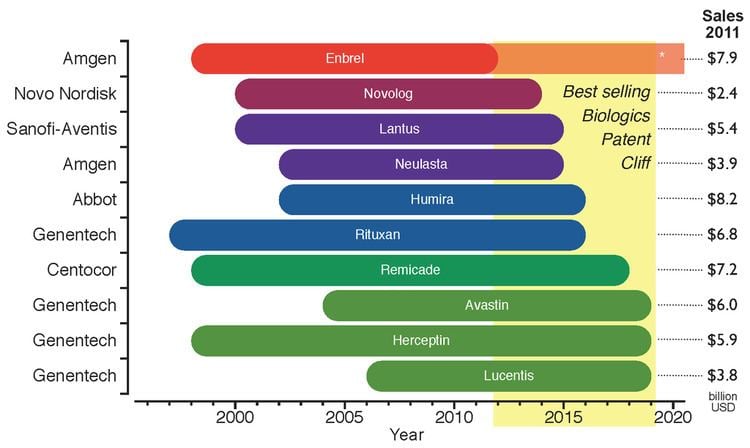
The legal requirements of approval pathways, together with the costly manufacturing processes, escalates the developing costs for biosimilars that could be between $75–$250 million per molecule. This market entry barrier affects not only the companies willing to produce them but could also delay availability of inexpensive alternatives for public healthcare institutions that subsidize treatment for their patients. Even though the biosimilars market is rising, the price drop for biological drugs at risk of patent expiration will not be as great as for other generic drugs; in fact it has been estimated that the price for biosimilar products will be 65%-85% of their originators. Biosimilars are drawing market's attention since there is an upcoming patent cliff, which will put nearly 36% of the $140 billion market for biologic drugs at risk (as of 2011), this considering only the top 10 selling products.
The global biosimilars market was $1.3 billion in 2013 and is expected to reach $35 billion by 2020 driven by the patent expiration of additional ten blockbuster biologic drugs.