
Entrez 158 | Ensembl ENSG00000239900 | |
 | ||
External IDs OMIM: 608222 MGI: 103202 HomoloGene: 12 GeneCards: ADSL | ||
Adenylosuccinate lyase (or adenylosuccinase) is an enzyme that in humans is encoded by the ADSL gene.
Contents
Adenylosuccinate lyase converts adenylosuccinate to AMP and fumarate as part of the purine nucleotide cycle. ASL catalyzes two reactions in the purine biosynthetic pathway that makes AMP; ASL cleaves adenylosuccinate into AMP and fumarate, and cleaves SAICAR into AICAR and fumarate.
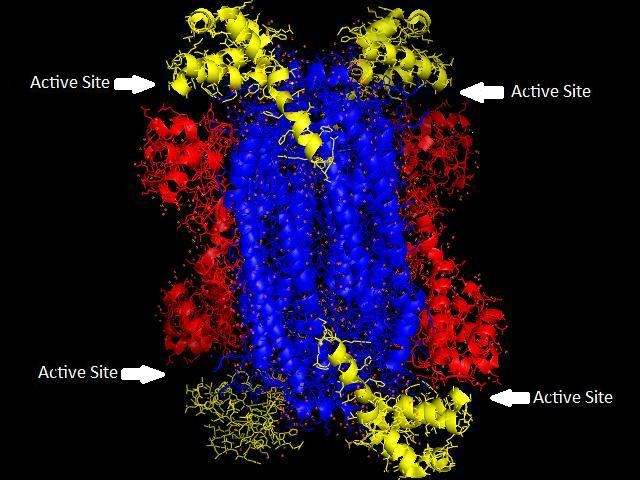
Adenylosuccinate lyase is part of the β-elimination superfamily of enzymes and it proceeds through an E1cb reaction mechanism. The enzyme is a homotetramer with three domains in each monomer and four active sites per homotetramer.
Point mutations in adenylosuccinate that cause lowered enzymatic activity cause clinical symptoms that mark the condition adenylosuccinate lyase deficiency.
This protein may use the morpheein model of allosteric regulation.
Function
Adenylosuccinate lyase (ASL) is an enzyme that catalyzes two reactions in the de novo purine biosynthetic pathway. In both reactions it uses an E1cb elimination reaction mechanism to cleave fumarate off of the substrate. In the first reaction, ASL converts 5-aminoimidazole- (N-succinylocarboxamide) ribotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribotide (AICAR) and fumarate. AICAR proceeds through three more reactions before it becomes adenylosuccinate (also called succinyladenosine monophosphate or SAMP), which ASL then splits into adenosine monophosphate (AMP) and fumarate. ASL is important to cells not only because of its involvement in creating purines needed for cellular replication, but also because it helps regulate metabolic processes by controlling the levels of AMP and fumarate in the cell.
Subunits
Adenylosuccinate lyase belongs to the β-elimination superfamily, and as such its structure is a homotetramer . The monomer of adenylosuccinate lyase has three domains. In Thermotoga maritima, domain 1 contains 7 α-helices in residues 1-93, including the His68 which is highly conserved and was previously thought to be the catalytic acid in the active site. More recent studies have posited that the His171 in domain 2, previously thought to be a catalytic base, may in fact be acting as the catalytic acid, at least in Escherichia coli. Domain 2 is made up of residues 94-341, and contains 5 α-helices and the monomer’s only β-sheet. Domain 3 is made up of 7 α-helices. The core of the tetramer is made up of the four domain 2 copies, and there are two copies each of domains 1 and 3 on each end of the tetramer giving the tetramer D2 dihedral symmetry. The tetramer has four active sites, each where three domains meet.
Adenylosuccinate lyase in humans and Bacillus subtilis can be competitively inhibited by the substrate analog adenosine phosphonobutyric acid 2’(3’), 5’-diphosphate (APBADP). APBADP is a competitive inhibitor for both of the reactions catalyzed by adenylosuccinate lyase, and kinetic studies with APBADP show that the substrates for both reactions use the same active site. In the ASL-catalyzed reaction splitting adenylosuccinate into adenosine monophosphate (AMP) and fumarate, the AMP must rotate slightly after the reaction is complete and before fumarate is released in order for both products to fit in the active site.
Mutations
Adenylosuccinate lyase mutants can have considerably reduced activity whether the mutation is in or away from the active site. Disease-causing ASL mutants R396C and R396H are at the entrance to the active site and have lower Vmax than the wild-type ASL, but the mutants K246E and L311V which are away from the active site also cause decreased Vmax. ASL mutant R194C is away from the active site, and though it maintains a Vmax similar to wild-type ASL, it was shown to be the least conformationally stable of the five mutants in vitro and still causes disease.
Mechanism
It was previously thought that the mechanism of action for adenylosuccinate lyase was a concerted catalysis where the hydrogen on the β-carbon (with respect to the leaving nitrogen) was abstracted by the catalytic base at the same time that the leaving nitrogen was protonated by the catalytic acid for E2 elimination. More recent data conflicts with this idea and has confirmed that the mechanism is not in fact concerted, but that the abstraction occurs first and there is an intermediate carbanion species which is resonance stabilized. For both ASL-catalyzed reactions deprotonation of the carbon β to the leaving nitrogen occurs first, then the formation and resonance stabilization of the carbanion occurs, and lastly the protonation of the leaving nitrogen which causes the C-N bond to break. Experimental confirmation of the deprotonation, carbanion formation, and the rate-limiting step of protonation causing cleavage means this is an E1cb mechanism. The most recent data suggest that the catalytic acid is His171, which was previously thought to be the catalytic base, and that somewhat unusually it is a serine at position 295 acts as the catalytic base. The cleavage of adenylosuccinate to AMP and fumarate is an ordered uni-bi mechanism, which means that after cleavage the fumarate leaves the active site before the AMP does.
Role in disease
Mutated adenylosuccinate lyase (ASL) causes clinical disease in patients that is referred to as adenylosuccinate lyase deficiency. This condition is rare, and it presents with varying degrees of psychomotor retardation, autism, muscle wasting, and epilepsy. The exact cause of disease is unknown, but possibilities include not enough purine nucleotide synthesis for cell replication, malfunctioning of the purine nucleotide cycle, and a buildup of substrates to toxic levels. Several disease-linked point mutations have been identified, and those who are heterozygous for a point mutation are healthy, but those who are homozygous develop clinical disease. The number of disease-causing genotypes keeps increasing as more mutations are discovered, and now thirty different point mutations have been identified so far, and one deletion, that cause adenylosuccinate lyase deficiency.
When the substrates of ASL (adenylosucinate and SAICAR) build up due to enzyme deficiency, they are dephosphorylated and turn into succinyladenosine (S-Ado) and succinylaminoimidazole carboximide riboside (SAICA riboside). Normally these compounds are not present in the cerebrospinal fluid or urine because ASL acts on the majority of the substrate molecules before they can build up and be phosphorylated. In the past there has not been a good test for adenylosuccinate lyase deficiency, making the rare disease difficult to diagnose, but recently a test was developed to detect SAICA and S-Ado in the urine. The test is inexpensive and had no false positives or false negatives in the researchers’ small sample.
It is thought that SAICA riboside may be the more toxic compound as it is found at higher levels in patients with severe clinical symptoms, and some researchers think S-Ado may even be protective. More research needs to be done on what determines disease severity, but the instability of human ASL in the lab setting has been an obstacle to this research.
Therapeutic applications
As resistance to anti-malarials increases, researchers are looking for new strategies to target the Plasmodium parasites which cause malaria, especially the more lethal P. falciparum. Some researchers suggested that ASL be looked into as a potential drug target because though interruption of the de novo purine biosynthesis pathway is toxic to the host, Plasmodium ASL has a low level of sequence homology with human ASL which may make any anti-Plasmodium ASL drugs specific enough not to harm human hosts.