
 | ||
Can vinyl cation be more favorable than allyl reactions of allene with hcl
Introduction, basics, and history
The vinyl cation is a carbocation with the positive charge on an alkene carbon. Its empirical formula is C2H3+. More generally, a vinylic cation is any disubstituted, trivalent carbon, where the carbon bearing the positive charge is part of a double bond and is sp hybridized. In the chemical literature, substituted vinylic cations are often referred to as vinyl cations, and understood to refer to the broad class rather than the C2H3+ variant alone. The vinyl cation is one of the main types of reactive intermediates involving a non-tetrahedrally coordinated carbon atom, and is necessary to explain a wide variety of observed reactivity trends. Vinyl cations are observed as reactive intermediates in solvolysis reactions, as well during electrophilic addition to alkynes, for example, through protonation of an alkyne by a strong acid. As expected from its sp hybridization, the vinyl cation prefers a linear geometry. Compounds related to the vinyl cation include allylic carbocations and benzylic carbocations, as well as aryl carbocations.
Contents
- Can vinyl cation be more favorable than allyl reactions of allene with hcl
- Introduction basics and history
- Structure of vinyl cations
- Stability of vinyl cations
- Electrophilic additions
- Rearrangements in vinyl cations
- Vinyl cations in pericyclic reactions
- Vinyl Cations in Hydrohalogenation
- References
Compared to other reactive intermediates such as radicals and carbanions, the vinyl cation long remained poorly-understood and were initially thought to be too high energy to form as reactive intermediates. Vinyl cations were first proposed in 1944 as a reactive intermediate for the acid-catalyzed hydrolysis of alkoxyacetylenes to give alkyl acetate. In the first step of their facile hydration reaction, which was the rate limiting step, a vinyl cation reactive intermediate was proposed; the positive charge was believed to formally lie on a diicoordinate carbon. This is the first time such a transition state can be found in the literature.
It wasn’t until the fifteen years later that this idea was revisited, with Grob and Cseh detecting vinyl cation formation during solvolysis reactions of alpha-vinyl halides in their seminal work. Indeed, for this contribution, Grob has been called “the father of the vinyl cation”. The 1960’s saw a flurry of vinyl cation-related research, with kinetics data driving the argument for the existence of the species. Noyce and coworkers, for example, reported the formation of a vinyl cation in acid-catalyzed hydration of phenylporopiolic acid. The authors note that in the rate limiting step, a large positive charge develops on the benzylic carbon, indicating that the reaction proceeds through a vinyl cation transition state. Hyperconjugation and hydrogen bonding was evoked to explain the accessibility of the vinyl cation described by Noyce.
Generating Vinyl Cations
Vinyl cations have been observed as reactive intermediates during solvolysis reactions. Consistent with SN1 chemistry, these reactions follow first order kinetics. Generally, vinylic halides are unreactive in solution: silver nitrate does not precipitate silver halides in the presence of vinyl halides, and this fact was historically used to dispute the existence of the vinyl cation species. The introduction of “super” leaving groups in the 1970’s first allowed for the generation of vinyl cation reactive intermediates with appreciable lifetimes. These excellent leaving groups, such as triflate (trifluoromethanesulfonate) and nonaflate (nonafluorobutanesulfonate), are highly prone to SN1 reactivity. Utilization of these super leaving groups allowed researchers for the first time to move beyond speculation about the existence of such vinyl cations.
Other leaving groups, such as hypervalent iodine moities (which are 1 million fold better leaving groups than the classic triflates), have been utilized to such end as well. Hinkle and coworkers synthesized a number of alkenyl(aryl)iodonium triflates from hypervalent phenyliodo precursors. In the scheme shown, the (E)- and (Z)- vinyl triflates form after heterolytic carbon-iodine bond cleavage and subsequent trapping of the cation by triflate. The presence of both (E)- and (Z)- vinyl triflate products offers support for the formation of a primary vinyl cation reactive intermediate; through SN2 chemistry, both only one isomer would form.
Recently, vinyl cation reactive intermediates have been generated in photochemical solvolysis reactions. The figure to the right depicts photochemical solvolysis of vinyl iodonium salt, through heterolytic carbon-iodine bond cleavage, to generate a vinyl carbocation and iodobenzene. The reactive intermediate is prone to either nucleophilic attack by the solvent to yield (E)- and (Z)- enol ether isomers, or beta hydrogen elimination.
Cyclic vinyl cations
The ease of generating cyclic vinyl cations depends on the size of the ring system, with vinyl cations residing on smaller rings being more difficult to produce. This trend is supported by calculations showing that the vinyl cation prefers a linear arrangement. Due to the high degree of strain in 3-membered ring systems, the generation of a the smallest cyclic vinyl cation, cycloprop-1-enyl cation, remains elusive. The SN1 solvolysis chemistry used to produce other vinyl cations has not proven facile for the cycloprop-1-enyl cation. This is a chemical challenge that remains unsolved.
Structure of vinyl cations
The simplest vinyl cation, C2H3+, which is unsubstituted, can have two possible structures, a classical linear or a non-classical bridged structure. Ab initio calculations have shown that the bridged structure is more stable than the classical by 5.0 kcal/mole. But for substituted vinyl cations with equivalent alkyl groups, the linear structure is supported by 13C and 1H NMR. The first experimental evidence of the linear structure of vinyl cations was the x-ray structure of b-silyl vinyl cations. Using multinuclear NMR spectroscopy, the compound exhibited a single 29Si NMR signal which implies that the two Si are equivalent and delocalize to the carbocation through hyperconjugation. The vinyl cation has an intense IR peak at 1987 cm−1 for the C=C+ stretching. More importantly, the bond angles between between the vinyl cation carbons and the first carbon of the alkyl substituted was measured to be approximately 180o.
Stability of vinyl cations
Initially it was believed that the existence of vinyl cations was questionable because of the large energy difference between it and its vinyl precursor. Once it was established that stable vinyl cation intermediates can be attained through the solvolysis of vinyl compounds with good leaving groups like triflate and nonaflate and stabilized by electron-donating groups, a significant amount of progress as taken place and produced a field of stable vinyl cations.
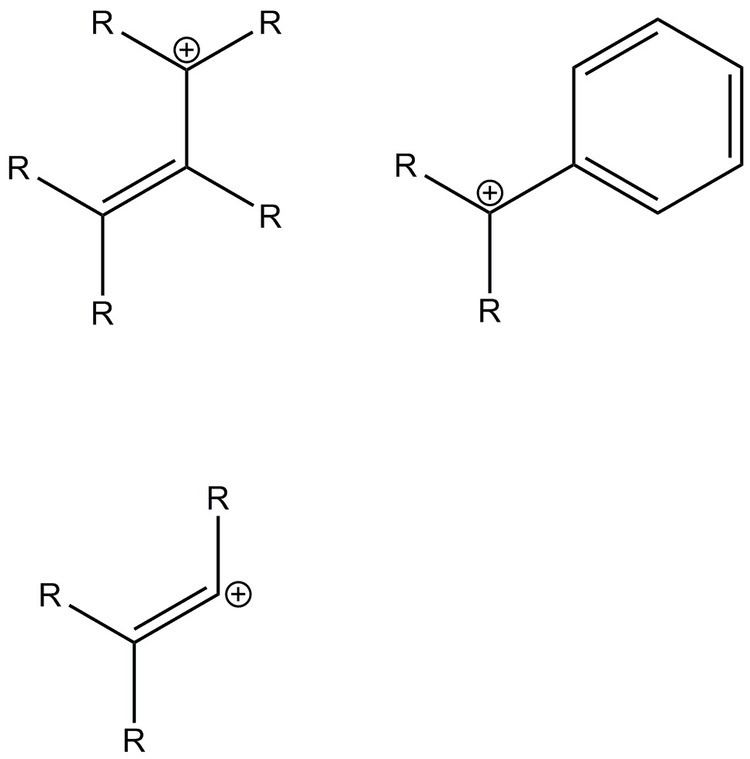
One of the earliest vinyl cations studied had aryl substituents with an electron-donating moiety. Arylvinyl compounds are stabilized by resonance. Upon the removal of the leaving group, the empty p-orbital is perpendicular to the conjugated system of the phenyl ring, so it can only achieve resonance stabilization in its transition state when the vinyl empty p-orbital is coplanar with the p system of the pnehyl ring. Adding steric bulk to the ortho-positions improve conjugation as it makes the phenyl ring orthogonal to the vinyl carbons but coplanar with the empty p-orbital.
Like arylvinyl cations, dienyl and allenyl cations are also stabilized by conjugation. Once again, double bonds in the conjugated system must be coplanar to the empty p-orbital to achieve resonance stabilization. In allenyl cations, the positive charge is well-distributed across the whole structure.
Cyclopropylvinyl cations exhibit a non-classical approach to stabilization. When it is in its bisected structure, there is suitable overlap between its empty p-orbital and the cyclopropyl ring that stabilization is achieved. In its other form, the perpendicular structure, the empty p-orbital is perpendicular to the ring system. The stabilizing power of the cyclopropyl ring is so great that it has become a driving thermodynamic force in rearrangements like 1,2-hydride shifts in (E) and (Z)-3-cyclopropyl-2-propenyl triflate solvolysis.
Substituent Effects on Vinyl Cation Stability
Table 1: Electronic effects responsible for stabilization of vinyl cation at the α-position
^ ‘-’ electron-withdrawing, ‘+’ electron-donating
# ‘+’ indicates stabilization and ‘-‘ indicates destabilization of the substituted vinyl cation with respect to neutral alkene equivalent
*indicates the strongest factor responsible for (de)stabilization for substituents that exhibit more than one electronic effect
** the substituent is inductively withdrawing at the carbonyl carbon and also exhibits small electron delocalization from the carbonyl oxygen
*** Y = -F, -Cl, -Br, -I, -OH, -CN, -CF3
The presence of an empty p-orbital perpendicular to the p-bond imparts unwanted destabilization onto the vinyl cation. This inherent instability can be diminished through favorable interactions with a-substituents that reduce the charge at the carbocation. Ab initio computational methods have been used to show stabilizing or destabilizing effects of substituents by monitoring changes in the enthalpies, bond lengths, bond order, and charges in the structures.
There are three possible electronic effects that a substituent may exhibit to influence the stability of the vinyl cation. It may either destabilize the cation by drawing even more electron density from the carbon or stabilizing by contributing more electron density. The carbocation positive charge can be relieved by the an unsaturated carbon-based or heteroatomic substituent through p-donation and/or C-H hyperconjugation by methylene/methyl substituents. In addition, inductive effects can either stabilize or destabilizing depending on whether the substituent is electron-donating or –withdrawing. Individual electronic effects are not isolable from the others as all three work together to influence the overall stability of the cation.
For vinyl cations, relative stabilities can be compared with respect to their neutral alkene analogs. To obtain the stabilization properties of a-substituents, the isodesmic reaction was used to calculate enthalpy differences between the substituted vinyl cation and its neutral alkene precursor by getting its reaction enthalpy. This method is advantageous as it can be benchmarked against experimentally-determined thermochemical values. Calculations are initialized from the bridged, nonclassical structure of vinyl cations as it is the global minimum.
In a preliminary work, 4 substituents (-CH=CH2, -F, -Cl, -CH3) were initially studied to investigate electronic effects on vinyl cation stability. Different a-substituents induces structural changes in the vinyl cation when compared to its neutral alkene counterpart. These changes can be attributed to the electronic effects present. In vinyl cations, there is a marked decrease in the C-R and C=C bond lengths, indicative of electron donation or induction between Ca and R, and Cb and Ca. On the other hand, the increase in the Cb-H bond length implies a strong hyperconjugative effect that is inversely related to the thermodynamic stability of the cation. Stabilization is possible because of a good overlap between the C-H bond and the empty p-orbital at Ca. Hyperconjugation is evident in all structures because of the adjacent Cb-H bond and in the –CH3 substituent.
Enthalpy calculations obtained from the isodesmic reaction are fair accurate and shows good correlation with experimental data. Stabilization is ranked the order, -F < -Cl < -CH3 < -CH=CH2. All substituents impart stability except for fluorine which destabilized the vinyl cation by 7 kcal/mole. This phenomenon can be explained by comparing a-fluorine substituent effects on vinyl and ethyl cations. In ethyl cations, fluorine stabilizes the carbocation. The stark difference in the stabilizing capabilities of fluorine in the vinyl and ethyl cation is due to the difference in the hybridization of the a-carbons. Because the vinyl cation has a more electronegative sp-hybridized carbon, inductive effects will be more prominent. Having electronegative sp-hybridized carbon interact with fluorine significantly destabilizes the structure. This phenomenon is also apparent in a lesser extent when comparing –CH3 and –CH=CH2 substituents, where -CH=CH2 is less stabilizing.
Heteroatoms like fluorine and chlorine, can exhibit both inductive (electron-withdrawing) and p-donation electronic effects because of their high electronegativities and p-electrons. Stabilization then depends on the balance between the two electronic effects. For fluorine, destabilization via induction is dominant and resonance is significantly weaker. While for chlorine, resonance is sufficient to counteract induction so that overall the effect is stabilizing.
For inductively withdrawing/donating and p-donating substituents, some partial charges reside in the R group and Ca. Although the trend in charge magnitude in R and Ca for the four substituents are inversely related. It is also observed that there is an increase in the bond order of Cb=Ca and and Ca-R, which is consistent with the corresponding changes in bond length.
In the small sample size of substituents, there was no observed correlation between bond order increase and charge distribution to R, and the stabilization due to the substituent. However, stabilization has exhibited a correlation to Cb-H bond elongation.
Based on the mechanisms provided above, a wide array of vinyl cation a-subtituents can be classified according to the electronic effects they exhibit and the extent of stabilization would depend on the delicate balance between these effects.
Lone pair-containing substituents like –NH2, -OH, and –SH are stabilizing since p-donation overcomes inductively withdrawing effects. Conjugated systems like –CH=CH2 and –C6H5 are stabilizing due to strong p-donation. Highly destabilizing substituents like –CF3 and –NO2 only exhibit inductive electron withdrawal. Weakly destabilizing substituents like –CN has a weak p-donation effect that does not completely curb induction by electron withdrawal.
It is not entirely plausible to isolate the inductive effect of heteroatomic a-substituents because other electronic effects get in the way. However, one way inductive effects of functional groups can be investigated is by probing b-substituent effects where the heteroatom would be a methylene group away from the vinyl cation (-CH2Y). In –CH2Y groups that exhibit a very small or no p donation, there is only a very small difference in the hyperconguative effect in the –CH2- groups of the substituents. Hence, the overall stability can be correlated to the b-substituent effect, now only driven by its inductive power. Comparing only purely inductively capabilities of functional groups the order is: CN > CF3 > F > Cl > Br > OH, with some destabilization energies comparable to a methyl group.
In most cases, substituents exhibit more than one electronic (de)stabilization effect. Usually, the inductive effect brought upon by multiple bonds to a heteroatom can be counterbalanced by p donation from the same heteroatom. For instance, based on absolute b-inductive power, -CN is more inductive than CF3, but since there can be p donation from the nitrogen of CN, its inductive capability is reduced. In common heteroatomic substituents like F, Cl, Br, and OH, the stabilization decreases with higher electron-withdrawing ability. However, p donation is still believed to take place because of C-R bond decrease.
Carbonyl substituents are mainly destabilizing because of the highly partially positive carbonyl carbon beside the vinyl cation and no p donation.
It is useful to compare substituent effects of vinyl cations and ethyl cations to investigate the hybridization effects of stabilization. In general, vinyl cations are more stabilized by substituents compared ethyl cations primarily because vinyl cations are inherently less stable to begin with. For strongly inductively electron-withdrawing groups like –F, -OH, and –NH2, inductive destablizaiton is more apparent in vinyl compared to ethyl cations because of the highly electronegative nature of vinyl cation sp hybrids compared to ethyl cation sp2 hybrids. In contrast, in the case of an α-Si(CH3)3 substituent, it is more stabilizing to vinyl cations because it has no p-electrons.
In terms of bond order, stabilizing substituents result in an increase in the C-R, Cα=Cβ, and Cβ-H bond orders. Small increases in bond orders are observed in –CF3, -CH2F, and –CH2X, where they are incapable of p donation, while large increases in bond orders are observed in substituents that can donate p or p electrons like –CH=CH2, -I, or –SH.
Electrophilic additions
A vinyl cation intermediate is possibly formed when electrophilic moieties attack unsaturated carbons. This can be achieved in the reaction of electrophiles with alkynes or allenes. In these reactions, a positive electrophile attacks on of the unsaturated carbons that then forms a vinyl cation, which subsequently undergoes further reaction steps to form the final product.
In the acid-catalyzed hydration of arylacetylene derivatives, a proton initially attacks the triple bond to form a vinyl cation at the aryl substituted carbon. The intermediate experiences little resonance stabilization because of the orthogonality of the congjuated aryl orbital with the empty p-orbital of the vinyl cation. The reaction is first order with respect to both the acetylene and the proton and the with the protonation of the acetylene as the rate-determining step. Monosubstituted aryl/alkoxy-acetylenes exhibit faster kinetics in acidic hydrations compared to its methyl-substituted equivalents. In arylacetylenes, methyl groups appear to contribute less stabilization compared to hydrogens because of C-H hyperconjugation, reversing the stabilization trend observed in alkyl cations. C-H hyperconjugation is a significant factor because the C-H bond can significantly overlap with the vacant p-orbital. Another possible explanation is that smaller size of the hydrogen substituent allows solvation to take place more easily contributing more significant stabilization.
Aside from protons, other electrophilic groups can attack an acetylene moiety. When attacked by carboxylic acids, cis/trans alkene adducts may be formed. The reaction with hydrogen halides, which also has an initial protonation step, results in the formation of halo-substituted alkenes. Lastly, adamantyl ketones may be formed from an adamantyl cation attack on acetylene and subsequent hydration.
In the hydrohalogenation of phenylpropene, two different alkene products are formed because of thermodynamic and kinetic effects. The linear sp-hybridized vinyl cation may be attacked by the halogen from two different directions. When attacked from the less sterically hindered side (hydrogen), the E-alkene is produced, attack to the other side forms the Z-alkene. Over short time scales, the E-alkene is favored because the attack from the less bulky side is preferred, but over longer times, the more stable (bulky methyl and phenyl groups on opposite sides) Z-alkene is preferred. Though the E-alkene is initially formed, it isomerizes to the Z-alkene through a carbocation intermediate the stems from protonation and C-C rotation steps.
Neighboring groups surround the alkyne can enhance reaction kinetics by interacting with the intermediate via nonclassical approaches like intramolecular interactions. An alkyne that is adjacent to a tertiary alcohol formas a four-membered cyclic vinyl cation intermediate in which the oxygen of the hydroxyl group bridges two carbons across two bonds. Likewise, a five-membered chloronium ring intermediate is formed from 5-chloro substituted 1-pentynes. An unusually shifted product is formed because the intermediate undergoes heterolysis at the C5-Cl position.
In the electrophilic attack of allenes, it takes place in a manner that prefers to form a terminal adduct and the vinyl cation at the central carbon. The polarization of the allene group show that the terminal carbons have a higher electron density and tendency to under nucleophillic attack. However, if the terminal end is stabilized by a substituent, an allyl-like cation may form as the electrophile attacks the central carbon. Similar to phenyl rings adjacent to vinyl cations, there must be bond rotation to achieve complete resonance stabilization.
Rearrangements in vinyl cations
Vinyl cations intermediates that are formed during reactions can have a tendency to undergo rearrangements. These rearrangements can be broadly categorized into to two classes: migrations into double bonds and rearrangements via the double bonds. The first category involves 1,2-shifts that lead to the formation of an allyl cation, while the second type involves the formation of another vinyl cation isomer.
Vinyl cations undergo 1,2-hydride shifts to form an allyl-stabilized cation. 1,2-hydride shifts are fairly common in alkyl cations and is fast in the NMR time scale. However, in vinyl cations, this rearrangement is uncommon even though the rearrangement product in thermodynamically stable. Much like the aryl-substituted vinyl cations, the interacting orbitals during the conversion of a linear vinyl cation to a non-linear allyl cation are orthogonal and passes through a non-planar transition state, which makes the rearrangement difficult. This is evident in the higher activation energies of 1,2-hydride shifts in vinyl cations compared to alkyl cations. Examples of reactions in which this is observed would be the protonation of di-alkyl substituted alkynes and in the solvolysis of ispropylvinyl trifluromethanesulfonate in trifluoroethanol.
1,2 methyl shifts also occurs in vinyl cations, and like 1,2-hydride shifts, it also has a higher activation barrier compared to its alkyl cation equivalent. In the protonation of alkynes, both 1,2-hydride and 1,2-methyl shifts may take place, the preference depends on the alkyl substituent since it will dictate the resulting allyl cation product. For t-butyl substituents, 1,2-methyl shifts are preferred, and for isopropyl substituents, 1,2-hydride shits occur instead. Cyclic alkenes also exhibit 1,2-methyl shifts upon solvolysis as well.
In the solvolysis of spiro-vinyl triflate, the formation of a vinyl cation intermediate through a concerted process drives further rearrangements that involve the formation of a completely different cyclic structure. Ring expansion can also be achieved through the rearrangement of a vinyl cation.
The second class of rearrangements, the vinyl cation rearranges to form another vinyl cation isomer. The process is highly dependent on the solvent, nature of the nucleophile, and moieties in the compound. In primary vinyl cations, a 1,2-hydride is unlikely because of the low stability of the primary vinyl cation because of the low electron-donating capability of hydrogen. However, this is still observed in special cases like in 1-methyl-2-phenylvinyl triflate, where the resulting vinyl cation is resonance-stabilized.
Methyl shifts are observed in the addition of tert-butyl cation to but-2-yne. The pentaallyl cation that is formed could be the result of a single 1,3-methyl shift or two consecutive 1,2-metyl shifts. Rearrangement via the double bond could also change the size of a cyclic system. In the solvolysis of methyl substituted cyclohexenyl triflate, the rearrangement and non-rearranged product are formed in almost equal amounts, with a small preference to the rearrangement product because of its linear structure. However, it must be noted that there is some strain in the methylenesyclopentane rearrangement product.
Lastly, halogens could also move into and stabilize a vinyl cation system. In the reaction of 5-chloropentyne with trifluoroacetic acid, there is simultaneous protonation and 1,4-shift of chlorine that forms a bridge cyclic structure across four carbons. Trifluoracetic acid subsequently attacks the intermediate from the terminal end to form 2-chloropent-4-enyl trifluoroacetate. This phenomena is also observed in other halogens. For instance, fluoroalkynes can form a product with two adducts.
Vinyl cations in pericyclic reactions
Ketenes and allenes undergo [2+2] cycloadditions under thermal conditions in a concerted manner because they have pi orbitals that are orthogonal to each other. Vinyl cation intermediates undergo the same process in the same manner because it has 2 p orbitals that can simultaneously overlap with the orbitals of the dienophile. In the Smirnov-Zamkow reaction between 2-butyne and Cl2, a cycloaddition leads to the formation of dichlorocyclobutane. A similar reaction is also observed when allene is reacted with HCl. After the cycloaddition, a cationic cyclic intermediate is formed and then it is attacked by a nucleophile to form the final product.
Vinyl Cations in Hydrohalogenation
There is debate on whether a vinyl cation intermediate forms with the addition of a halide (H-X) compound to a terminal alkyne for hydrohalogenation reactions. Alternatively, some believe that the addition of H and Br in this case is actually concerted.