
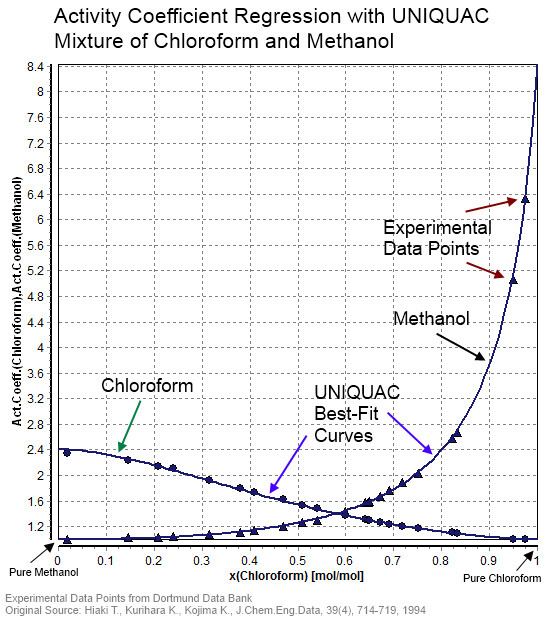
 | ||
UNIQUAC (short for UNIversal QUAsiChemical) is an activity coefficient model used in description of phase equilibria. The model is a so-called lattice model and has been derived from a first order approximation of interacting molecule surfaces in statistical thermodynamics. The model is however not fully thermodynamically consistent due to its two liquid mixture approach. In this approach the local concentration around one central molecule is assumed to be independent from the local composition around another type of molecule.
Contents
- Equations
- Combinatorial contribution
- Residual contribution
- Applications
- Parameters
- Newer developments
- References
The UNIQUAC model can be considered a second generation activity coefficient because its expression for the Excess Gibbs energy consists of an entropy term in addition to an enthalpy term. Earlier activity coefficient models such as the Wilson equation and the Non-random two-liquid model (NRTL model) only consist of enthalpy terms.
Today the UNIQUAC model is frequently applied in the description of phase equilibria (i.e. liquid–solid, liquid–liquid or liquid–vapor equilibrium). The UNIQUAC model also serves as the basis of the development of the group contribution method UNIFAC, where molecules are subdivided into functional groups. In fact, UNIQUAC is equal to UNIFAC for mixtures of molecules, which are not subdivided; e.g. the binary systems water-methanol, methanol-acryonitrile and formaldehyde-DMF.
A more thermodynamically consistent form of UNIQUAC is given by the more recent COSMOSPACE and the equivalent GEQUAC model.
Equations
In the UNIQUAC model the activity coefficients of the ith component of a two component mixture are described by a combinatorial and a residual contribution.
The first is an entropic term quantifying the deviation from ideal solubility as a result of differences in molecule shape. The latter is an enthalpic correction caused by the change in interacting forces between different molecules upon mixing.
Combinatorial contribution
The combinatorial contribution accounts for shape differences between molecules and affects the entropy of the mixture and is based on the lattice theory. The excess entropy γC is calculated exclusively from the pure chemical parameters, using the relative Van der Waals volumes ri and surface areas qi of the pure chemicals.
With the volume fraction per mixture mole fraction, Vi, for the ith component given by:
And the surface area fraction per mixture molar fraction, Fi, for the ith component given by:
The first 3 terms on the right hand side of the combinatorial term form the Flory-Huggins contribution, while the left terms, the Guggenhem-Staverman correction, reduce this because a connecting segments cannot be placed in all direction in space. This spatial correction shifts the result of the Flory-Huggins term about 5% towards an ideal solution. The coordination number, z, i.e. the number of close interacting molecules around a central molecule, is frequently set to 10. It can be regarded as an average value that lies between cubic (z=6) and hexagonal packing (z=12) of molecules that are simplified by spheres.
In the limit of infinite dilution and a binary mixture the equations for the combinatorial contribution reduces to:
This pair of equation shows that molecules of same shape, i.e. same r and q parameters, have
Residual contribution
The residual, enthalpic term contains an empirical parameter,
with
Δuij [J/mol] is the binary interaction energy parameter. Theory defines Δuij = uij – uii, and Δuji = uji – ujj, where uij is the interaction energy between molecules i and j. The interaction energy parameters are usually determined from activity coefficients, vapor-liquid, liquid-liquid, or liquid-solid equilibrium data.
Usually Δuij ≠ Δuji, because the energies of evaporation (i.e. uii), are in many cases different, while the energy of interaction between molecule i and j is symmetric, and therefore uij=uji. If the interactions between the j molecules and i molecules is the same as between molecules i and j, than mixing has no excess energy effect upon mixing, Δuij=Δuji=0. And thus
Alternatively, in some process simulation software
The "C", "D", and "E" coefficients are primarily used in fitting liquid–liquid equilibria (with "D" and "E" rarely used at that). The "C" coefficient is useful in vapor-liquid equilibria as well. The use of such an expression ignores the fact that on a molecular level the energy, Δuij, is temperature independent. It is a correction to repair the simplifications, which were applied in the derivation of the model.
Applications
Activity coefficients can be used to predict simple phase equilibria (vapour–liquid, liquid–liquid, solid–liquid), or to estimate other physical properties (e.g. viscosity of mixtures). Models such as UNIQUAC allow chemical engineers to predict the phase behavior of multicomponent chemical mixtures. They are commonly used in process simulation programs to calculate the mass balance in and around separation units.
Parameters
UNIQUAC requires two basic underlying parameters:
- Relative surface and volume fractions are chemical constants, which must be known for all chemicals.
- An empirical parameter between components that describes the intermolecular behaviour. This parameter must be known for all binary pairs in the mixture. In a quaternary mixture there are six such parameters (1–2,1–3,1–4,2–3,2–4,3–4) and the number rapidly increases with additional chemical components. The empirical parameters are derived from experimental activity coefficients, or from phase diagrams, from which the activity coefficients themselves can be calculated. An alternative is to obtain activity coefficients with a method such as UNIFAC, and the UNIFAC parameters can then be simplified by fitting to obtain the UNIQUAC parameters. This method allows for the more rapid calculation of activity coefficients, rather than direct usage of the more complex method.
Newer developments
UNIQUAC has been extended by several research groups. Some selected derivatives are: