
 | ||
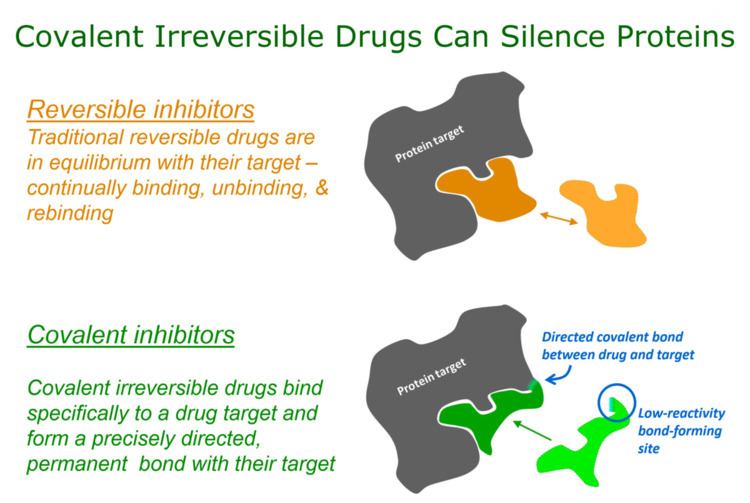
Targeted covalent inhibitors (TCIs) or Targeted covalent drugs are rationally designed inhibitors that bind and then bond to their target proteins. These inhibitors possess a bond-forming functional group of low chemical reactivity that, following binding to the target protein, is positioned to react rapidly with a specific nucleophilic residue at the target site and form a bond (Singh, Petter, Baillie, & Whitty, 2011a).
Contents
- Historical impact of covalent drugs
- Potency
- Selectivity
- Pharmacodynamics
- Built in biomarker
- Design of covalent drugs
- Toxicity risks associated with covalent modification of proteins
- TCIs in Clinical Development
- EGFR and Lung Cancer
- ErbB Family and Breast Cancer
- Btk and Leukemia
- Covalent drug companies
- Publications by Academic Groups Working on Covalent Inhibitors
- References
Historical impact of covalent drugs
Over the last 100 years covalent drugs have made a major impact on human health and have been highly successful drugs for the pharmaceutical industry (Robertson, 2005)). These inhibitors react with their target proteins to form a covalent complex in which the protein has lost its function. The majority of these successful drugs, which include penicillin, omeprazole, clopidogrel, and aspirin were discovered through serendipity in phenotypic screens (Potashman & Duggan, 2009).
For the past 20 years key changes in screening approaches, along with safety concerns, have made pharma reluctant to pursue covalent inhibitors in a systematic way (Liebler & Guengerich, 2005)(Park et al., 2011).
Recently, there has been considerable attention to using rational drug design to create highly selective covalent inhibitors called targeted covalent inhibitors (Singh, Petter, & Kluge, 2010). The first published example of a targeted covalent drug was for the EGFR kinase (Singh et al., 1997) (Fry et al., 1998) but this has now broadened to other kinases (Singh et al., 2010) (Cohen, Zhang, Shokat, & Taunton, 2005) and other protein families (Hagel et al., 2011; Nacht et al., 2010) (Ostrem, Peters, Sos, Wells, & Shokat, 2013).
Potency
Covalent bonding can lead to potencies and ligand efficiencies that are either exceptionally high or, for irreversible covalent interactions, even essentially infinite. Covalent bonding thus allows high potency to be routinely achieved in compounds of low molecular mass, along with all the beneficial pharmaceutical properties that are associated with small size (Smith, Zhang, Leach, & Houk, 2009)
Selectivity
Covalent inhibitors can be designed to target a nucleophile that is unique or rare across a protein family (Singh et al., 1997)(Cohen et al., 2005)(Singh et al., 2010)(Liu et al., 2013), thereby ensuring that covalent bond formation cannot occur with most other family members. This approach can lead to high selectivity against closely related proteins because although the inhibitor might bind transiently to the active sites of such proteins, it will not covalently label them if they lack the targeted nucleophilic residue in the appropriate position.
Pharmacodynamics
The restoration of pharmacological activity after covalent irreversible inhibition requires re-synthesis of the protein target. This has important and potentially advantageous consequences for drug pharmacodynamics in which the level and frequency of dosing relates to the extent and duration of the resulting pharmacological effect(Durham & Blanco, 2015).
Built-in-biomarker
Covalent inhibitors can be used to assess target engagement which can sometimes be used pre-clinically and clinically to assess the relationship between dose of drug and efficacy or toxicity (Durham & Blanco, 2015). This approach was used for covalent Btk inhibitors pre-clinically and clinically to understand the relationship between dose administered and efficacy in animal models of arthritis and target occupancy in a clinical study of healthy volunteers (Evans et al., 2011)(Advani et al., 2013).
Design of covalent drugs
The design of covalent drugs requires careful optimization of both the non-covalent binding affinity (which is reflected in Ki) and the reactivity of the electrophilic warhead (which is reflected in k2).
The initial design of TCIs* involves three key steps. First, bioinformatics analysis is used to identify a nucleophilic amino acid (for example, cysteine) that is either inside or near to a functionally relevant binding site on a drug target, but is rare in that protein family. Next, a reversible inhibitor is identified for which the binding mode is known. Finally, structure-based computational methods are used to guide the design of modified ligands that have electrophilic functionality, and are positioned to react specifically with the nucleophilic amino acid in the target protein (Singh et al., 2011a)
Toxicity risks associated with covalent modification of proteins
There has been a reluctance for modern drug discovery programs to consider covalent inhibitors due to toxicity concerns (Park et al., 2011). An important contributor has been the drug toxicities of several high-profile drugs believed to be caused by metabolic activation of reversible drugs(Park et al., 2011). For example, high dose acetaminophen can lead to the formation of the reactive metabolite N-acetyl-p-benzoquinone imine. Also, covalent inhibitors such as beta lactam antibiotics which contain weak electrophiles can lead to idiosyncratic toxicities (IDT) in some patients. It has been noted that many approved covalent inhibitors have been used safely for decades with no observed idiosyncratic toxicity. Also, that IDTs are not limited to proteins with a covalent mechanism of action(Bauer, 2015). A recent analysis has noted that the risk of idiosyncratic toxicities may be mitigated through lower doses of administered drug. Doses of less than 10 mg per day rarely lead to IDT irrespective of the drug mechanism(Nakayama et al., 2009).
TCIs in Clinical Development
Despite the apparent lack of attention towards covalent inhibitor drug discovery by most pharmaceutical companies, there are several examples of covalent drugs that have been approved or are progressing to late-stage clinical development.
EGFR and Lung Cancer
The second generation EGFR inhibitor Afatinib has been approved for the treatment of EGFR driven lung cancer and Dacomitinib is in late stage clinical testing. The third generation EGFR inhibitors which target mutant EGFR which is specific to the tumor but are selective against wild-type EGFR that are expected to lead to a wider therapeutic index (Chong, P. Janne, 2012). Rociletinib has published early clinical data showing progress in mutant driven EGFR lung cancer [1]. Also, AZD9291 has made considerable clinical progress [Jänne PA, et al. A Phase I study of AZD9291 in patients with EGFR-TKI-resistant advanced NSCLC – updated progression-free survival and duration of response data. Presented at the European Lung Cancer Conference (ELCC) Annual Meeting, Geneva; 15–18 April 2015] in EGFR driven lung cancer patients.
ErbB Family and Breast Cancer
There has also been progress in the treatment of metastatic breast cancer with the irreversible inhibitor Neratinib which targets the ErbB family of tyrosine kinases. Neratinib is in multiple Phase 2 and also a Phase 3 clinical trial for the treatment of breast cancer].
Btk and Leukemia
Ibrutinib, a covalent inhibitor of Bruton's tyrosine kinase, has been approved for the treatment of chronic lymphocytic leukemia, waldenstrom’s macroglobulinemia and mantle cell lymphoma.