
 | ||
Single-cell DNA template strand sequencing, or strand-seq, is a technique for the selective sequencing of a daughter cell’s parental template strands. This technique has many applications, including the identification of sister chromatid exchanges in the parental cell prior to segregation, the identification of misoriented contigs during alignment of reads to the reference genome, and the assessment of non-random segregation of sister chromatids.
Contents
- Background
- Similar methods
- Pulse chase
- Chromosome orientation fluorescence in situ hybridization CO FISH
- Wet lab protocols
- Bioinformatic processing
- Limitations
- Identifying sister chromatid exchanges
- Identifying misoriented contigs
- Identifying non random segregation of sister chromatids
- Identifying aneuploid chromosomes
- Genome assembly
- Considerations
- References
Background
Strand-seq (single strand sequencing) was first described in 2012 as a technique to isolate sequenced reads from parental template strands in single-cell DNA libraries. As a proof of concept study, the authors demonstrated the ability to acquire sequence information from the Watson and/or Crick chromosomal strands in an individual DNA library, depending on the mode of chromatid segregation; a typical DNA library will always contain DNA from both strands. The authors were specifically interested in showing the utility of strand-seq in detecting sister chromatid exchanges (SCEs) at high-resolution. They successfully identified eight putative SCEs in the murine (mouse) embryonic stem (meS) cell line with resolution up to 23 bp. This methodology has also been shown to hold great utility in discerning patterns of non-random chromatid segregation, especially in stem cell lineages. Furthermore, SCEs have been implicated as diagnostic indicators of genome stress, information that has utility in cancer biology. Most research on this topic involves observing the assortment of chromosomal template strands through many cell development cycles and correlating non-random assortment with particular cell fates. Single-cell sequencing protocols were foundational in the development of this technique, but they differ in several aspects.
Similar methods
Past methods have been used to track the inheritance patterns of chromatids on a per-strand basis and elucidate the process of non-random segregation:
Pulse-chase
Pulse-chase experiments have been used for determining the segregation patterns of chromosomes in addition to studying other time-dependent cellular processes. Briefly, pulse-chase assays allow researchers to track radioactively labelled molecules in the cell. In experiments used to study non-random chromosome assortment, stem cells are labeled or "pulsed" with a nucleotide analog that is incorporated in the replicated DNA strands. This allows the nascent stands to be tracked through many rounds of replication. Unfortunately, this method is found to have poor resolution as it can only be observed at the chromatid level.
Chromosome-orientation fluorescence in situ hybridization (CO-FISH)
CO-FISH, or strand-specific fluorescence in situ hybridization, facilitates strand-specific targeting of DNA with fluorescently-tagged probes. It exploits the uniform orientation of major satellites relative to the direction of telomeres, thus allowing strands to be unambiguously designated as "Watson" or "Crick" strands. Using unidirectional probes that recognize major satellite regions, coupled to fluorescently labelled dyes, individual strands can be bound. To ensure that only the template strand is labelled, the newly formed strands must be degraded by BrdU incorporation and photolysis. This protocol offers improved cytogenetic resolution, allowing researchers to observe single strands as opposed to whole chromatids with pulse-chase experiments. Moreover, non-random segregation of chromatids can be directly assayed by targeting major satellite markers.
Wet lab protocols
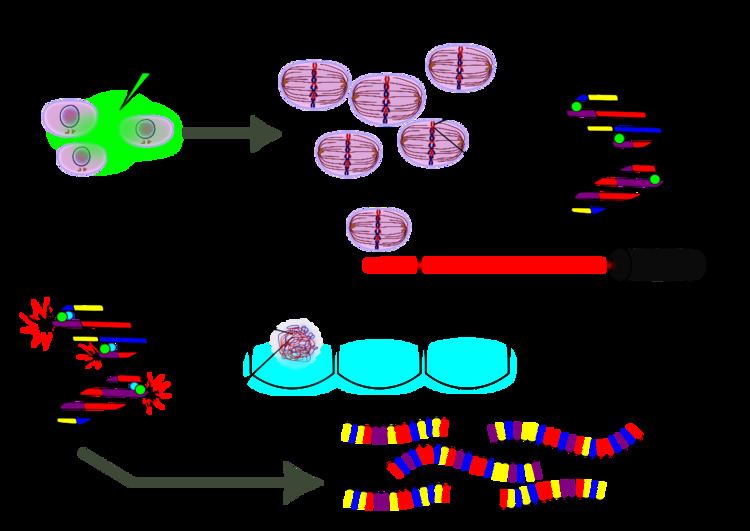
Cells of interest are cultured either in vivo or in vitro. During S-phase cells are treated with bromodeoxyuridine (BrdU) which is then incorporated into their nascent DNA, acting as a substitute for thymidine. After at least one replication event has occurred, the daughter cells are synchronized at the G2 phase and individually separated by fluorescence-activated cell sorting (FACS). The cells are directly sorted into lysis buffer and their DNA is extracted. Having been arrested at a specified number of generations (usually one), the inheritance patterns of sister chromatids can be assessed. The following methods concentrate on the DNA sequencing of a single daughter cell’s DNA. At this point the chromosomes are composed of nascent strands with BrdU in place of thymidine and the original template strands are primed for DNA sequencing library preparation. Since this protocol was published in 2012, the canonical methodology is only well described for Illumina sequencing platforms; the protocol could very easily be adapted for other sequencing platforms, depending on the application. Next, the DNA is incubated with a special dye such that when the BrdU-dye complex is excited by UV light, nascent strands are nicked by photolysis. This process inhibits polymerase chain reaction (PCR) amplification of the nascent strand, allowing only the parental template strands to be amplified. Library construction proceeds as normal for Illumina paired-end sequencing. Multiplexing PCR primers are then ligated to the PCR amplicons with hexamer barcodes identifying which cell each fragment they are derived from. Unlike single cell sequencing protocols, Strand-seq does not utilize multiple displacement amplification or MALBAC for DNA amplification. Rather, it is solely dependent on PCR.
Bioinformatic processing
The majority of current applications for Strand-seq start by aligning sequenced reads to a reference genome. Alignment can be performed using a variety of short-read aligners such as BWA and Bowtie. By aligning Strand-seq reads from a single cell to the reference genome, the inherited template strands can be determined. If the cell was sequenced after more than one generation, a pattern of chromatid assortment can be ascertained for the particular cell lineage at hand. Presently, the Bioinformatic Analysis of Inherited Templates (BAIT) is the only bioinformatic software to exclusively analyze reads generated from the Strand-seq methodology. It begins by aligning the reads to a reference sequence, binning the genome into sections, and finally counting the number of Watson and Crick reads falling within each bin. From here, BAIT enables the identification of SCE events, misoriented contigs in the reference genome, aneuploid chromosomes and modes of sister chromatid segregation. It can also aid in assembling early-build genomes and assigning orphan scaffolds to locations within late-build genomes.
Limitations
The protocols have been published for only the Illumina HiSeq sequencing platform using paired-end sequencing. Applications that require sequence information from different sequencing technologies would require new protocols. Authors from the papers describing Strand-seq showed that they were able to attain a 23bp resolution for mapping SCEs. Other large chromosomal abnormalities would likely share that mapping resolution. This may be dependent on a combination of the sequencing platform used, library preparation protocols, and the number of cells analysed. More experimentation will be necessary to discover the root of this inaccuracy. However, it would be sensical for precision to increase with sequencing technologies that don’t incur errors in homopolymeric repeats and by including more cells in analyses.
Identifying sister chromatid exchanges
Strand-seq was initially proposed as a tool to identify sister chromatid exchanges. Being a process that is localized to individual cells, DNA sequencing of more than one cell would naturally scatter these effects and suggest an absence of SCE events. Moreover, classic single cell sequencing techniques are unable to show these events due to heterogeneous amplification biases and dual-strand sequence information, thereby necessitating Strand-seq. Using the reference alignment information, researchers can identify an SCE if the directionality of an inherited template strand changes.
Identifying misoriented contigs
Misoriented contigs are present in reference genomes at significant rates (ex. 1% in the mouse reference genome). Strand-seq, in contrast to conventional sequencing methods, can detect these misorientations. Misoriented contigs are present where strand inheritance changes from one homozygous state to the other (ex. WW to CC, or CC to WW). Moreover, this state change is visible in every Strand-seq library, reinforcing the presence of a misoriented contig.
Identifying non-random segregation of sister chromatids
Prior to the 1960s, it was assumed that sister chromatids were segregated randomly into daughter cells. However, non-random segregation of sister chromatids has been observed in mammalian cells ever since. There have been a few hypotheses proposed to explain the non-random segregation, including the Immortal Strand Hypothesis and the Silent Sister Hypothesis, one of which may hopefully be verified by methods involving Strand-seq.
‘’Immortal Strand Hypothesis’’
Mutations occur every time a cell divides. Certain long-lived cells (ex. stem cells) may be particularly affected by these mutations. The Immortal Strand Hypothesis proposes that these cells avoid mutation accumulation by consistently retaining parental template strands[9]. For this hypothesis to be true, sister chromatids from each and every chromosome must segregate in a non-random fashion. Additionally, one cell will retain the exact same set of template strands after each division, giving the rest to the other cell products of the division.
‘’Silent Sister Hypothesis’’
This hypothesis states that sister chromatids have differing epigenetic signatures, thereby also differing expression regulation. When replication occurs, non-random segregation of sister chromatids ensures the fates of the daughter cells. Assessing the validity of this hypothesis would require a joint analysis of Strand-seq and gene expression profiles for both daughter cells.
Identifying aneuploid chromosomes
The output of BAIT shows the inheritance of parental template strands. Normally, two template strands are inherited for each chromosome, and any deviation from this number indicates an instance of aneuploidy.
Genome assembly
Early-build genomes are quite fragmented, with unordered and unoriented contigs. Using Strand-seq provides directionality information to accompany the sequence, which ultimately helps resolve the placement of contigs. Contigs present in the same chromosome will exhibit the same directionality, provided SCE events have not occurred. Conversely, contigs present in different chromosomes will only exhibit the same directionality in 50% of the Strand-seq libraries. Scaffolds, successive contigs intersected by a gap, can be localized in the same manner.
Considerations
The possibility that BrdU being substituted for thymine in the genomic DNA could induce double stranded chromosomal breaks and specifically resulting in SCEs has been previously discussed in the literature. Additionally, BrdU incorporation has been suggested to interfere with strand segregation patterns. If this is the case, there would be an inflation in false positive SCEs which may be annotated. Therefore, many cells should be analyzed using the Strand-seq protocol to ensure that SCEs are in fact present in the population. The number of single cell strands that need to be sequenced in order for an annotation to be accepted has yet to be proposed and is highly dependent on the questions being asked. As Strand-seq is founded on single cell sequencing techniques, one must consider the problems faced with single cell sequencing as well. These include the lacking standards for cell isolation and amplification. Even though previous Strand-seq studies isolated cells using FACS, microfluidics also serves as an attractive alternative. PCR has been shown to produce more erroneous amplification products compared to strand displacement based methods such as MDA and MALBAC. Strand displacement amplification also tends to generate more sequence and longer products which could be beneficial for long read sequencing technologies.