
 | ||
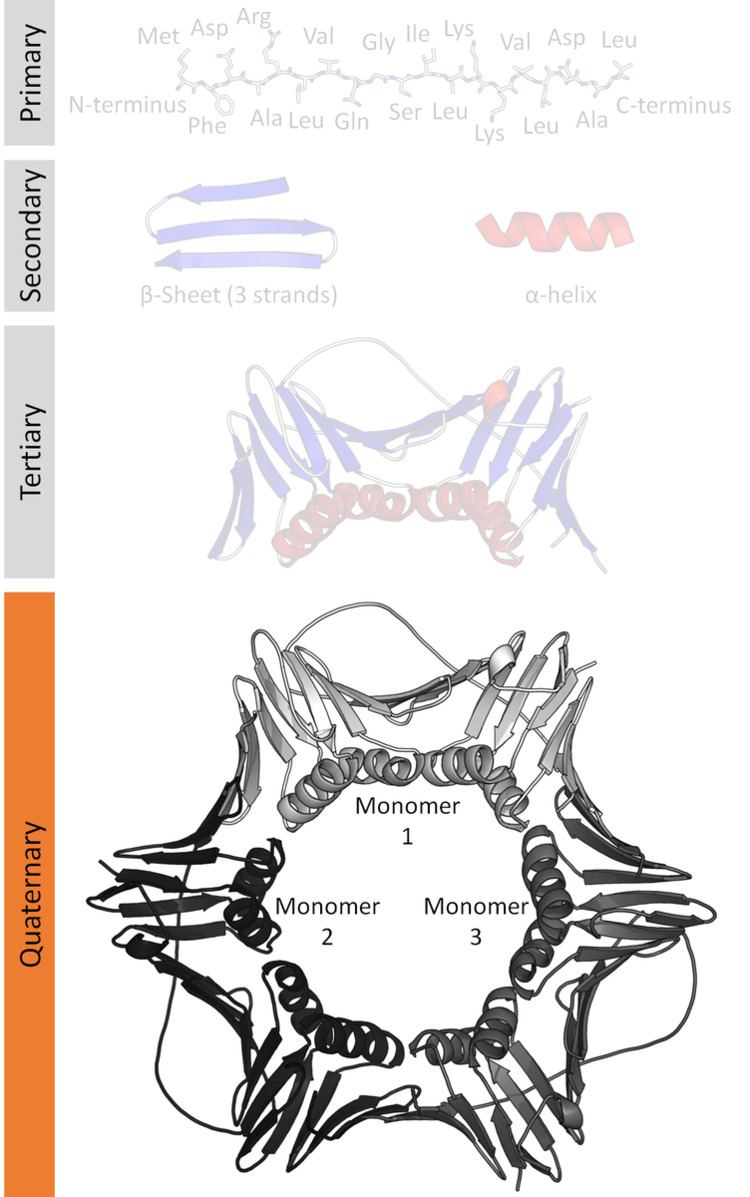
Protein quaternary structure is the number and arrangement of multiple folded protein subunits in a multi-subunit complex. It includes organisations from simple dimers to large homooligomers and complexes with defined or variable numbers of subunits. It can also refer to biomolecular complexes of proteins with nucleic acids and other cofactors.
Contents
Description and examples
Many proteins are actually assemblies of multiple polypeptide chains. The quaternary structure refers to the number and arrangement of the protein subunits with respect to one another. Examples of proteins with quaternary structure include hemoglobin, DNA polymerase, and ion channels.
Enzymes composed of subunits with diverse functions are sometimes called holoenzymes, in which some parts may be known as regulatory subunits and the functional core is known as the catalytic subunit. Other assemblies referred to instead as multiprotein complexes also possess quaternary structure. Examples include nucleosomes and microtubules. Changes in quaternary structure can occur through conformational changes within individual subunits or through reorientation of the subunits relative to each other. It is through such changes, which underlie cooperativity and allostery in "multimeric" enzymes, that many proteins undergo regulation and perform their physiological function.
The above definition follows a classical approach to biochemistry, established at times when the distinction between a protein and a functional, proteinaceous unit was difficult to elucidate. More recently, people refer to protein–protein interaction when discussing quaternary structure of proteins and consider all assemblies of proteins as protein complexes.
Nomenclature
The number of subunits in an oligomeric complex is described using names that end in -mer (Greek for "part, subunit"). Formal and Greco-Latinate names are generally used for the first ten types and can be used for up to twenty subunits, whereas higher order complexes are usually described by the number of subunits, followed by -meric.
*No known examplesAlthough complexes higher than octamers are rarely observed for most proteins, there are some important exceptions. Viral capsids are often composed of multiples of 60 proteins. Several molecular machines are also found in the cell, such as the proteasome (four heptameric rings = 28 subunits), the transcription complex and the spliceosome. The ribosome is probably the largest molecular machine, and is composed of many RNA and protein molecules.
In some cases, proteins form complexes that then assemble into even larger complexes. In such cases, one uses the nomenclature, e.g., "dimer of dimers" or "trimer of dimers", to suggest that the complex might dissociate into smaller sub-complexes before dissociating into monomers.
Determination
Protein quaternary structure can be determined using a variety of experimental techniques that require a sample of protein in a variety of experimental conditions. The experiments often provide an estimate of the mass of the native protein and, together with knowledge of the masses and/or stoichiometry of the subunits, allow the quaternary structure to be predicted with a given accuracy. It is not always possible to obtain a precise determination of the subunit composition for a variety of reasons.
The number of subunits in a protein complex can often be determined by measuring the hydrodynamic molecular volume or mass of the intact complex, which requires native solution conditions. For folded proteins, the mass can be inferred from its volume using the partial specific volume of 0.73 ml/g. However, volume measurements are less certain than mass measurements, since unfolded proteins appear to have a much larger volume than folded proteins; additional experiments are required to determine whether a protein is unfolded or has formed an oligomer.
Prediction
Some bioinformatics methods were developed for predicting the quaternary structural attributes of proteins based on their sequence information by using various modes of pseudo amino acid composition (see, e.g., refs.).
Direct mass measurement of intact complexes
Direct size measurement of intact complexes
Indirect size measurement of intact complexes
Methods that measure the mass or volume under unfolding conditions (such as MALDI-TOF mass spectrometry and SDS-PAGE) are generally not useful, since non-native conditions usually cause the complex to dissociate into monomers. However, these may sometimes be applicable; for example, the experimenter may apply SDS-PAGE after first treating the intact complex with chemical cross-link reagents.
Protein–protein interactions
Proteins are capable of forming very tight complexes. For example, ribonuclease inhibitor binds to ribonuclease A with a roughly 20 fM dissociation constant. Other proteins have evolved to bind specifically to unusual moieties on another protein, e.g., biotin groups (avidin), phosphorylated tyrosines (SH2 domains) or proline-rich segments (SH3 domains).