
eMedicine radio/458 | ||
 | ||
ICD-10 Q61.4 (EUROCAT Q61.40-Q61.41) | ||
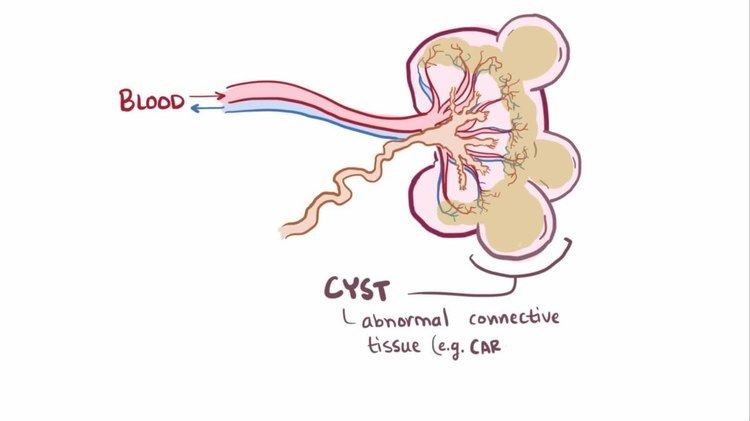
Multicystic dysplastic kidney (MCDK) is a condition that results from the malformation of the kidney during fetal development. The kidney consists of irregular cysts of varying sizes. Multicystic dysplastic kidney is a common type of renal cystic disease, and it is a cause of an abdominal mass in infants.
Contents
Signs and symptoms
When a diagnosis of multicystic kidney is made in utero by ultrasound, the disease is found to be bilateral in many cases. Those with bilateral disease often have other severe deformities or polysystemic malformation syndromes. In bilateral cases, the newborn has the classic characteristic of Potter's syndrome.
The bilateral condition is incompatible with survival, as the contralateral system frequently is abnormal as well. Contralateral ureteropelvic junction obstruction is found in 3% to 12% of infants with multicystic kidney and contralateral vesicoureteral reflux is seen even more often, in 18% to 43% of infants. Because the high incidence of reflux, voiding cystourethrography usually has been considered advisable in all newborns with a multicystic kidney.
Cause
The cause of multicystic dysplastic kidney can be attributed to genetics.Renal dysplasia can be a consequence of a genetic syndrome, which in turn may affect the digestive tract, nervous system, or other areas of the urinary tract. If the mother had been taking certain prescription drugs such as those for hypertension, this may be a precipitating factor as well.
Pathophysiology
The mechanism of multicystic dysplastic kidney is a result of an abnormal induction of metanephric mesenchyme. This could be a result of a formation difficulty of the mesonephric duct. Some mutations in genes associated with renal dysplasia (in syndromes)have been determined. The mutations in question occur at EYA1 or SIX1 genes (branchio-oto-renal syndrome). The PAX2 gene is also thought to play a role in MCDK.
Diagnosis
MCDK is usually diagnosed by ultrasound examination before birth. Mean age at the time of antenatal diagnosis is about 28 weeks A microscopic analysis of urine in individuals with probable multicystic dysplastic kidney should be done. One meta-analysis demonstrated that unilateral MCDK occurs more frequently in males and the greater percentage of MCKD occur on the left side of the body.
Treatment
MCDK is not treatable. However, the patient is observed periodically for the first few years during which ultrasounds are generally taken to ensure the healthy kidney is functioning properly and that the unhealthy kidney is not causing adverse effects. In severe cases MCDK can lead to neonatal fatality (in bilateral cases), however in unilateral cases the prognosis might be better (it would be dependent on associated anomalies).
Epidemiology
In regard to the epidemiology of multicystic dysplasia kidney, the incidence of MCDK is estimated to be 1 in every 4,000 live births, making it rare in terms of the general population.