
Symbol MCCC1 HUGO 6936 RefSeq NM_020166 | Entrez 56922 OMIM 609010 UniProt Q96RQ3 | |
 | ||
3 methylcrotonyl coa carboxylase deficiency medical condition
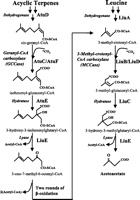
Methylcrotonyl CoA carboxylase (MCC) (3-methylcrotonyl CoA carboxylase, methylcrotonoyl-CoA carboxylase) is a biotin-requiring enzyme located in the mitochondria. MCC uses bicarbonate as a carboxyl group source to catalyze the carboxylation of a carbon adjacent to a carbonyl group performing the fourth step in processing leucine, an essential amino acid.
Contents
- 3 methylcrotonyl coa carboxylase deficiency medical condition
- Gene
- Protein
- Function
- Mechanism
- Regulation
- Clinical significance
- Interactions
- References
Gene
Human MCC is a biotin dependent mitochondrial enzyme formed by the two subunits MCCCα and MCCCβ, encoded by MCCC1 and MCCC2 respectively. MCCC1 gene has 21 exons and resides on chromosome 3 at q27. MCCC2 gene has 19 exons and resides on chromosome 5 at q12-q13.
Protein
The enzyme contains α and β subunits. Human MCCCα is composed of 725 amino acids which harbor a covalently bound biotin essential for the ATP-dependent carboxylation; MCCCβ has 563 amino acids that possess carboxyltransferase activity which presumably is essential for binding to 3-methylcrotonyl CoA. The MCC holoenzyme is thought to be a heterododecamer (6α6β) with close structural analogy to propionyl-CoA carboxylase (PCC), another biotin dependent mitochondrial carboxylase.
Function
During branched-chain amino acid degradation, MCC performs a single step in the breakdown of leucine to eventually yield acetyl CoA and acetoacetate. MCC catalyzes the carboxylation of 3-methylcrotonyl CoA to 3-methylglutaconyl CoA, a critical step for leucine and isovaleric acid catabolism in species including mammals, plants and bacteria. 3-Methylglutaconyl CoA is then hydrated to produce 3-hydroxy-3-methylglutaryl CoA. 3-Hydroxy-3-methylglutaryl CoA is cleaved into two molecules, acetoacetate and acetyl CoA.
Point mutations and deletion events in the genes coding for MCC can lead to MCC deficiency, an inborn error of metabolism which usually presents with vomiting, metabolic acidosis, very low plasma glucose concentration, and very low levels of carnitine in plasma.
Mechanism
Bicarbonate is activated by the addition of ATP, increasing the reactivity of bicarbonate. Once bicarbonate is activated, the biotin portion of MCC performs nucleophilic attack on the activated bicarbonate to form enzyme-bound carboxybiotin. The carboxybiotin portion of MCC can then undergo nucleophilic attack transferring the carboxyl group to the substrate, 3-methylcrotonyl CoA, to form 3-methylglutaconyl CoA.
Regulation
MCC is not regulated by small molecules or dietary or hormonal factors.
Clinical significance
In humans, MCC deficiency is a rare autosomal recessive genetic disorder whose clinical presentations range from benign to profound metabolic acidosis and death in infancy. Defective mutations in either the α or β subunit have been shown to cause the MCC-deficient syndrome. The typical diagnostic test is the elevated urinary excretion of 3-hydroxyisovaleric acid and 3-methylcrotonylglycine. Patients with MCC deficiency usually have normal growth and development before the first acute episode, such as convulsions or coma, that usually occurs between the age of 6-months to 3-years.
Interactions
MCC has been shown to interact with TRI6 in Fusarium graminearum.