
Trade names Prexige Routes ofadministration Oral Molar mass 293.72 g/mol | Pregnancycategory AU: C ATC code M01AH06 (WHO) CAS ID 220991-20-8 | |
 | ||
AHFS/Drugs.com International Drug Names | ||
How to pronounce lumiracoxib
Lumiracoxib (rINN) is a carboxylic acid COX-2 selective inhibitor non-steroidal anti-inflammatory drug, manufactured by Novartis and still sold in few countries, including Mexico, Ecuador and the Dominican Republic, under the trade name Prexige (sometimes misquoted as "Prestige" by the media).
Contents

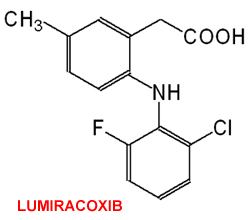
Lumiracoxib has several distinctive features. Its structure is different from that of other COX-2 inhibitors, such as celecoxib: lumiracoxib is an analogue of diclofenac (one chlorine substituted by fluorine, the phenylacetic acid has another methyl group in meta position), making it a member of the arylalkanoic acid class of NSAIDs; it binds to a different site on the COX-2 enzyme than do other COX-2 inhibitors; it is the only acidic coxib and has the highest COX-2 selectivity of any NSAID.

Since its original approval, lumiracoxib has been withdrawn from the market in several countries, mostly due to its potential for causing liver failure (sometimes requiring liver transplantation). It has never been approved for use in the United States.

History
The TARGET study (Therapeutic Arthritis Research and Gastrointestinal Event Trial) was conducted with more than 18,000 patients to test its gastrointestinal and cardiovascular safety against naproxen and ibuprofen and also study its efficacy against these two NSAIDs.
In November 2006, Prexige received marketing approval for all European Union countries through a common procedure called MRP. However, in August 2007, Prexige was withdrawn from the market in Australia following 8 serious liver adverse events, including 2 deaths and 2 liver transplants. On September 27, 2007, the US Food and Drug Administration issued a not approvable letter for lumiracoxib, requiring additional safety data. Canada withdrew Prexige (approved at 100 mg dose only) in October 2007. Several European Union countries followed suit in November 2007.

The FDA rejected Prexige as a trade name for lumiracoxib in 2003. Prexede was suggested as an alternative, but the FDA Division of Medication Errors and Technical Support (DMETS) subsequently recommended against it as well.
Withdrawal from market

On August 11, 2007, Australia's Therapeutic Goods Administration (TGA, the national agency responsible for regulation of pharmaceuticals) cancelled the registration of lumiracoxib in Australia due to concerns that it may cause liver failure.
According to the TGA's Principal Medical Adviser, Dr Rohan Hammett, as of 10 August 2007 the TGA had received 8 reports of serious adverse liver reactions to the drug, including two deaths and two liver transplants.
"The TGA and its expert advisory committee, the Adverse Drug Reactions Advisory Committee (ADRAC), have urgently investigated these reports. ADRAC has today recommended the cancellation of the registration of Lumiracoxib due to the severity of the reported side effects associated with this drug," Dr Hammett said.
"The TGA has taken this advice to cancel the registration of Lumiracoxib in order to prevent further cases of severe liver damage.
"It seems that the longer people are on the medicine, the greater the chance of liver injury. The TGA is, therefore, advising people to stop taking the Lumiracoxib immediately and to discuss alternative treatments with their doctor," Dr Hammett said.
New Zealand has followed suit with Australia in recalling Prexige.
On October 3, 2007, Health Canada requested sales of Prexige to stop. Novartis has agreed to the request and has taken steps to do so. On December 13, 2007, the European Medicines Agency recommended the withdrawal for Prexige from all EU markets.
On January 17, 2008, the Philippines Department of Health ordered Novartis Healthcare Phils. Inc. (Novartis) to remove (recall) all lumiracoxib from local drug stores in 2 weeks due to the harmful effects of the drug (potential serious liver-related side effects, hepatotoxicity or malfunction of the lungs).
On July 22, 2008, The Brazilian National Health Surveillance Agency ordered the withdrawal of 100 mg formulations of lumiracoxib and suspended marketing of the 400 mg formulation for 90 days, after a three-year safety review found a marked increase in adverse event reports; 35% of lumiracoxib-associated adverse events reported worldwide between July 2005 and April 2008 were found to have occurred in Brazil. Lumiracoxib was definitively withdrawn from the Brazilian market on October 3, 2008.
On November 12, 2008, INVIMA, the Colombian National Institute for Food and Drug Surveillance ordered the withdrawal of all presentations of lumiracoxib (Prexige), due to the international reports on hepatotoxicity.