
 | ||
In phylogenetics, long branch attraction (LBA) is a form of systematic error whereby distantly related lineages are incorrectly inferred to be closely related. LBA arises when the amount of molecular or morphological change accumulated within a lineage is sufficient to cause that lineage to appear similar (thus closely related) to another long-branched lineage, solely because they have both undergone a large amount of change, rather than because they are related by descent. Such bias is more common when the overall divergence of some taxa results in long branches within a phylogeny. Long-branches are often attracted to the base of a phylogenetic tree, because the lineage included to represent an outgroup is often also long-branched. The frequency of true LBA is unclear and often debated. Although often viewed as a failing of parsimony-based methodology, LBA can result from a variety of scenarios and be inferred under multiple analysis paradigms.
Contents
Causes
LBA was first recognized as problematic when analyzing discrete morphological character sets under parsimony criteria, however Maximum Likelihood analyses of DNA or protein sequences are also susceptible. A simple hypothetical example can be found in Felsenstein 1978 where it is demonstrated that for certain unknown "true" trees, some methods can show bias for grouping long branches, ultimately resulting in the inference of a false sister relationship. Often this is because convergent evolution of one or more characters included in the analysis has occurred in multiple taxa. Although they were derived independently, these shared traits can be misinterpreted in the analysis as being shared due to common ancestry.
In phylogenetic and clustering analyses, LBA is a result of the way clustering algorithms work: terminals or taxa with many autapomorphies (character states unique to a single branch) may by chance exhibit the same states as those on another branch (homoplasy). A phylogenetic analysis will group these taxa together as a clade unless other synapomorphies outweigh the homoplastic features to group together true sister taxa.
These problems may be minimized by using methods that correct for multiple substitutions at the same site, by adding taxa related to those with the long branches that add additional true synapomorphies to the data, or by using alternative slower evolving traits (e.g. more conservative gene regions).
Results
The result of LBA in evolutionary analyses is that rapidly evolving lineages may be inferred to be closely related, regardless of their true relationships. For example, in DNA sequence-based analyses, the problem arises when sequences from two (or more) lineages evolve rapidly. There are only four possible nucleotides and when DNA substitution rates are high, the probability that two lineages will evolve the same nucleotide at the same site increases. When this happens, parsimony may erroneously interpret this homoplasy as a synapomorphy (i.e., evolving once in the common ancestor of the two lineages).
The opposite effect may also be observed, in that if two (or more) branches exhibit particularly slow evolution among a wider, fast evolving group, those branches may be misinterpreted as closely related. As such, "long branch attraction" can in some ways be better expressed as "branch length attraction". However, it is typically long branches that exhibit attraction.
The recognition of long-branch attraction implies that there is some other evidence that suggests that the phylogeny is incorrect. For example, morphological data may suggest that taxa marked as closely related are not truly sister taxa. Hennig's Auxiliary Principle suggests that synapomorphies should be viewed as de facto evidence of grouping unless there is specific contrary evidence (Hennig, 1966; Schuh and Brower, 2009).
A simple and effective method for determining whether or not long branch attraction is affecting tree topology is the SAW method, named for Siddal and Whiting. If long branch attraction is suspected in a pair of taxa (A and B), simply remove taxon A ("saw" off the branch) and re-run the analysis. Then remove A and replace B, running the analysis again. If either of the taxa appear at different branch points in the absence of the other, there is evidence of long branch attraction. Since long branches can't possibly attract one another when only one is in the analysis, consistent taxon placement between treatments would indicate long branch attraction is not a problem.
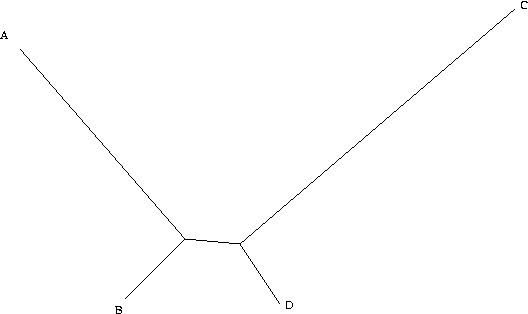
Example
Assume for simplicity that we are considering a single binary character (it can either be + or -). Because the distance from B to D is small, in the vast majority of all cases, B and D will be the same. Here, we will assume that they are both + (+ and - are assigned arbitrarily and swapping them is only a matter of definition). If this is the case, there are four remaining possibilities. A and C can both be +, in which case all taxa are the same and all the trees have the same length. A can be + and C can be -, in which case only one character is different, and we cannot learn anything, as all trees have the same length. Similarly, A can be - and C can be +. The only remaining possibility is that A and C are both -. In this case, however, we group A and C together, and B and D together. As a consequence, when we have a tree of this type, the more data we collect (i.e. the more characters we study), the more we tend towards the wrong tree.