
 | ||
Hydroamination is the addition of an N-H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries.
Contents
- History
- Reaction scope
- Products
- Catalysts
- Catalytic cycles
- Thermodynamics and kinetics
- Thermodynamic vs kinetic product
- Base catalyzed hydroamination
- Hydroamination catalyzed by group IV complexes
- Formal hydroamination
- Applications
- References
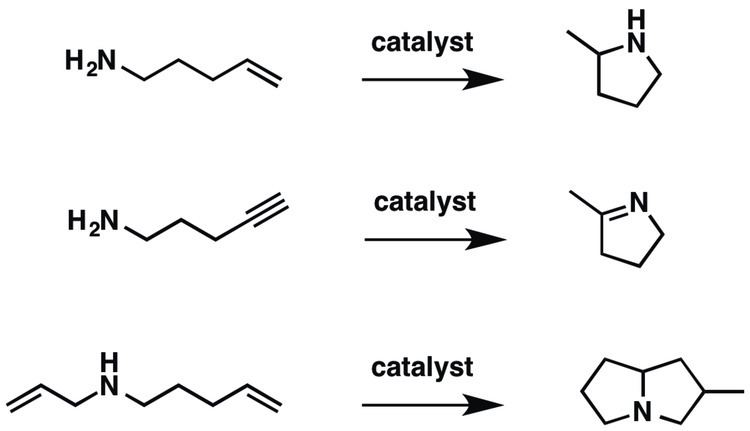
Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes.
History
The first intramolecular hydroaminations were reported by Tobin J. Marks in 1989 using metallocene derived from rare-earth metals such as lanthanum, lutetium, and samarium. Catalytic rates correlated inversely with the ionic radius of the metal, perhaps as a consequence of steric interference from the ligands. In 1992, Marks developed the first chiral hydroamination catalysts by using a chiral auxiliary, which were the first hydroamination catalysts to favor only one specific stereoisomer. Chiral auxiliaries on the metallocene ligands were used to dictate the stereochemistry of the product. The first non-metallocene chiral catalysts were reported in 2003, and used bisarylamido and aminophenolate ligands to give higher enantioselectivity.
Reaction scope
Hydroamination has been examined with a variety of amines, unsaturated substrates, and vastly different catalysts. Amines that have been investigated span a wide scope including primary, secondary, cyclic, acyclic, and anilines with diverse steric and electronic substituents. The unsaturated substrates that have been investigated include alkenes, dienes, alkynes, and allenes. For intramolecular hydroamination, various aminoalkenes have been examined.
Products
Addition across the unsaturated carbon-carbon bond can be Markovnikov or anti-Markovnikov depending on the catalyst. Interestingly, when considering the possibly of R/S chirality, four products can be obtained: Markovnikov with R or S and anti-Markovnikov addition with R or S. Although there have been many reports of catalytic hydroamination with a wide range of metals, there are far fewer describing enantioselective catalysis to selectively make one of the four possible products. Recently, there have been reports of selectively making the thermodynamic or kinetic product, which can be related to the racemic Markovnikov or anti-Markovnikov structures (see Thermodynamic and Kinetic Product below).
Catalysts
Many metal-ligand combinations have been reported to catalyze hydroamination, including main group elements including alkali metals such as lithium, group 2 metals such as calcium, as well as group 3 metals such as aluminum, indium, and bismuth. In addition to these main group examples, extensive research has been conducted on the transition metals with reports of early, mid, and late metals, as well as first, second, and third row elements. Finally the lanthanides have been thoroughly investigated. Zeolites have also shown utility in hydroamination.
Catalytic cycles
The mechanism of metal-catalyzed hydroamination has been well studied. Particularly well studied is the organolanthanide catalyzed intramolecular hydroamination of alkenes. First, the catalyst is activated by amide exchange, generating the active catalysis (i). Next, the alkene inserts into the Ln-N bond (ii). Finally, protonolysis occurs generating the cyclized product while also regenerating the active catalyst (iii). Although this mechanism depicts the use of a lanthanide catalyst, it is the basis for rare-earth, actinide, and alkali metal based catalysts.
Late transition metal hydroamination catalysts have multiple models based on the regioselective determining step. The four main categories are (1) nucleophilic attack on an alkene alkyne, or allyl ligand and (2) insertion of the alkene into the metal-amide bond. Generic catalytic cycles appear below. Mechanisms are supported by rate studies, isotopic labeling, and trapping of the proposed intermediates.
Thermodynamics and kinetics
The hydroamination reaction is approximately thermochemically neutral. The reaction however suffers from a high activation barrier, perhaps owing to the repulsion of the electron-rich substrate and the amine nucleophile. The intermolecular reaction also is accompanied by highly negative changing entropy, making it unfavorable at higher temperatures. Consequently, catalysts are necessary for this reaction to proceed. As usual in chemistry, intramolecular processes occur at faster rates than intermolecular versions.
Thermodynamic vs kinetic product
In general, most hydroamination catalysts require elevated temperatures to function efficiently, and as such, only the thermodynamic product is observed. The isolation and characterization of the rarer and more synthetically valuable kinetic allyl amine product was reported when allenes was used at the unsaturated substrate. One system utilized temperatures of 80 °C with a rhodium catalyst and aniline derivatives as the amine. The other reported system utilized a palladium catalyst at room temperature with a wide range of primary and secondary cyclic and acyclic amines. Both systems produced the desired allyl amines in high yield, which contain an alkene that can be further functionalized through traditional organic reactions.
Base catalyzed hydroamination
Strong bases catalyze hydroamination, an example being the ethylation of piperidine using ethylene:
Such base catalyzed reactions proceed well with ethylene but higher alkenes are less reactive.
Hydroamination catalyzed by group (IV) complexes
Certain titanium and zirconium complexes catalyze intermolecular hydroamination of alkynes and allenes. Both stoichiometric and catalytic variants were initially examined with zirconocene bis(amido) complexes. Titanocene amido and sulfonamido complexes catalyze the intra-molecular hydroamination of aminoalkenes via a [2+2] cycloaddition that forms the corresponding azametallacyclobutane, as illustrated in Figure 1. Subsequent protonolysis by incoming substrate gives the α-vinyl-pyrrolidine (1) or tetrahydropyridine (2) product. Experimental and theoretical evidence support the proposed imido intermediate and mechanism with neutral group IV catalysts.
Formal hydroamination
The addition of hydrogen and an amino group (NR2) using reagents other than the amine HNR2 is known as a "formal hydroamination" reaction. Although the advantages of atom economy and/or ready available of the nitrogen source are diminished as a result, the greater thermodynamic driving force, as well as ability to tune the aminating reagent are potentially useful. In place of the amine, hydroxylamine esters and nitroarenes have been reported as nitrogen sources.
Applications
Hydroamination could find applications due to the valuable nature of the resulting amine, as well as the greenness of the process. Functionalized allylamines, which can be produced through hydroamination, have extensive pharmaceutical application, although presently such species are not prepared by hydroamination. Hydroamination has been utilized to synthesize the allylamine Cinnarizine in quantitative yield. Cinnarizine treats both vertigo and motion sickness related nausea.
Hydroamination is also promising for the synthesis of alkaloids. An example was the total synthesis of (-)-epimyrtine.