
Specialty medical genetics ICD-9-CM 526.89, 733.29,756.54 eMedicine radio/284 | ICD-10 K10.8, M85.0, Q78.1 MedlinePlus 001234 MeSH D005357 | |
 | ||
Fibrous dysplasia is a disorder where normal bone and marrow is replaced with fibrous tissue, resulting in formation of bone that is weak and prone to expansion. As a result, most complications result from fracture, deformity, functional impairment, and pain. Disease occurs along a broad clinical spectrum ranging from asymptomatic, incidental lesions to severe disabling disease. Disease can affect one bone (monostotic) or multiple (polyostotic), and may occur in isolation or in combination with cafe-au-lait skin macules and hyperfunctioning endocrinopathies, termed McCune-Albright syndrome. More rarely, fibrous dysplasia may be associated with intramuscular myxomas, termed Mazabraud's syndrome. Fibrous dysplasia is very rare, and there is no known cure. Fibrous dysplasia is not a form of cancer.
Contents
Pathophysiology
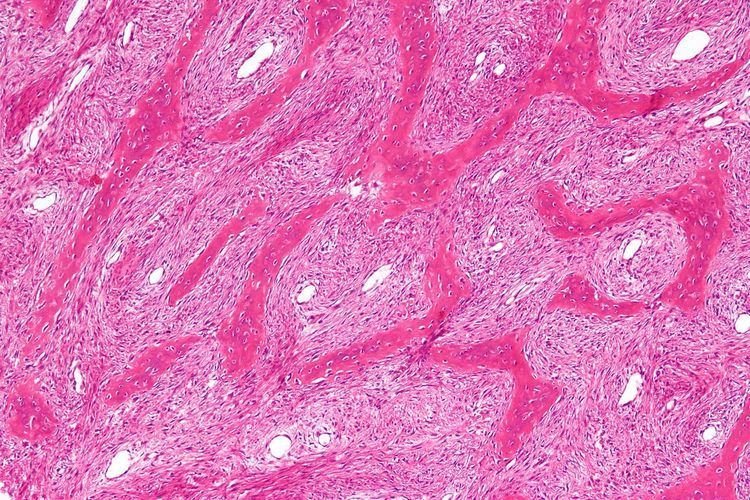
Fibrous dysplasia is a mosaic disease resulting from post-zygotic activating mutations of the GNAS locus at 20q13.2-q13.3, which codes for the α subunit of the Gs G-coupled protein receptor. In bone, constitutive Gsα signaling results in impaired differentiation and proliferation of bone marrow stromal cells. Proliferation of these cells causes replacement of normal bone and marrow with fibrous tissue. The bony trabeculae are abnormally thin and irregular, and often likened to Chinese characters (bony spicules on biopsy).
Fibrous dysplasia is not hereditary, and there has never been a case of transmission from parent to child.
Presentation
Fibrous dysplasia is a mosaic disease that can involve any part or combination of the craniofacial, axillary, and/or appendicular skeleton. The type and severity of the complications therefore depend on the location and extent of the affected skeleton. The clinical spectrum is very broad, ranging from an isolated, asymptomatic monostotic lesion discovered incidentally, to severe disabling disease involving practically the entire skeleton and leading to loss of vision, hearing, and/or mobility.
Individual bone lesions typically manifest during the first few years of life and expand during childhood. The vast majority of clinically significant bone lesions are detectable by age 10 years, with few new and almost no clinically significant bone lesions appearing after age 15 years. Total body scintigraphy is useful to identify and determine the extent of bone lesions, and should be performed in all patients with suspected fibrous dysplasia.
Children with fibrous dysplasia in the appendicular skeleton typically present with limp, pain, and/or pathologic fractures. Frequent fractures and progressive deformity may lead to difficulties with ambulation and impaired mobility. In the craniofacial skeleton, fibrous dysplasia may present as a painless “lump” or facial asymmetry. Expansion of craniofacial lesions may lead to progressive facial deformity. In rare cases patients may develop vision and/or hearing loss due to compromise of the optic nerves and/or auditory canals, which is more common in patients with McCune-Albright syndrome associated growth hormone excess. Fibrous dysplasia commonly involves the spine, and may lead to scoliosis, which in rare instances may be severe. Untreated, progressive scoliosis is one of the few features of fibrous dysplasia that can lead to early fatality.
Bone pain is a common complication of fibrous dysplasia. It may present at any age, but most commonly develops during adolescence and progresses into adulthood.
Bone marrow stromal cells in fibrous dysplasia produce excess amounts of the phosphate-regulating hormone fibroblast growth factor-23 (FGF23), leading to loss of phosphate in the urine. Patients with hypophosphatemia may develop rickets/osteomalacia, increased fractures, and bone pain.
Treatment
Treatment in fibrous dysplasia is mainly palliative, and is focused on managing fractures and preventing deformity. There are no medications capable of altering the disease course. Intravenous bisphosphonates may be helpful for treatment of bone pain, but there is no clear evidence that they strengthen bone lesions or prevent fractures. Surgical techniques that are effective in other disorders, such as bone grafting, curettage, and plates and screws, are frequently ineffective in fibrous dysplasia and should be avoided. Intramedullary rods are generally preferred for management of fractures and deformity in the lower extremities. Progressive scoliosis can generally be managed with standard instrumentation and fusion techniques. Surgical management in the craniofacial skeleton is complicated by frequent post-operative FD regrowth, and should focus on correction of functional deformities. Prophylactic optic nerve decompression increases the risk of vision loss and is contraindicated.
Managing endocrinopathies is a critical component of management in FD. All patients with fibrous dysplasia should be evaluated and treated for endocrine diseases associated with McCune–Albright syndrome. In particular untreated growth hormone excess may worsen craniofacial fibrous dysplasia and increase the risk of blindness. Untreated hypophosphatemia increases bone pain and risk of fractures.