
DiseasesDB 32380 | eMedicine neuro/556 | |
 | ||
OMIM 160120 108500 606554 606552 | ||
Episodic ataxia (EA) is an autosomal dominant disorder characterized by sporadic bouts of ataxia (severe discoordination) with or without myokymia (continuous muscle movement). There are seven types recognised but the majority are due to two recognized entities. Ataxia can be provoked by stress, startle, or heavy exertion such as exercise. Symptoms can first appear in infancy. There are at least 6 loci for EA, of which 4 are known genes. Some patients with EA also have migraine or progressive cerebellar degenerative disorders, symptomatic of either familial hemiplegic migraine or spinocerebellar ataxia. Some patients respond to acetazolamide though others do not.
Contents
Signs/Symptoms
Typically, episodic ataxia presents as bouts of ataxia induced by startle, stress, or exertion. Some patients also have continuous tremors of various motor groups, known as myokymia. Other patients have nystagmus, vertigo, tinnitus, diplopia or seizures.
Cause
The various symptoms of EA are caused by dysfunction of differing areas. Ataxia, the most common symptom, is due to misfiring of Purkinje cells in the cerebellum. This is either due to direct malfunction of these cells, such as in EA2, or improper regulation of these cells, such as in EA1. Seizures are likely due to altered firing of hippocampal neurons (KCNA1 null mice have seizures for this reason).
EA1: KCNA1
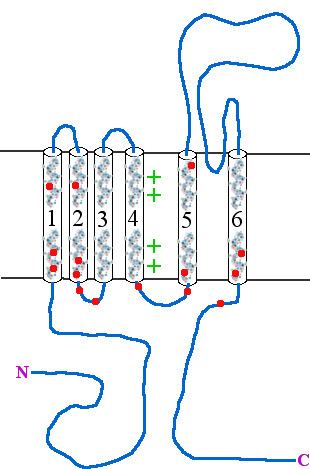
Type 1 episodic ataxia (EA1) is characterized by attacks of generalized ataxia induced by emotion or stress, with myokymia both during and between attacks. This disorder is also known as episodic ataxia with myokymia (EAM), hereditary paroxysmal ataxia with neuromyotonia and Isaacs-Mertens syndrome. Onset of EA1 occurs during early childhood to adolescence and persists throughout the patient's life. Attacks last from seconds to minutes. Mutations of the gene KCNA1, which encodes the voltage-gated potassium channel KV1.1, are responsible for this subtype of episodic ataxia. KV1.1 is expressed heavily in basket cells and interneurons that form GABAergic synapses on Purkinje cells. The channels aid in the repolarization phase of action potentials, thus affecting inhibitory input into Purkinje cells and, thereby, all motor output from the cerebellum. EA1 is an example of a synaptopathy. There are currently 17 KV1.1 mutations associated with EA1, Table 1 and Figure 1. 15 of these mutations have been at least partly characterized in cell culture based electrophysiological assays wherein 14 of these 15 mutations have demonstrated drastic alterations in channel function. As described in Table 1, most of the known EA1 associated mutations result in a drastic decrease in the amount of current through KV1.1 channels. Furthermore, these channels tend to activate at more positive potentials and slower rates, demonstrated by positive shifts in their V½ values and slower τ activation time constants, respectively. Some of these mutations, moreover, produce channels that deactivate at faster rates (deactivation τ), which would also result in decreased current through these channels. While these biophysical changes in channel properties likely underlie some of the decrease in current observed in experiments, many mutations also seem to result in misfolded or otherwise mistrafficked channels, which is likely to be the major cause of dysfunction and disease pathogenesis. It is assumed, though not yet proven, that decrease in KV1.1 mediated current leads to prolonged action potentials in interneurons and basket cells. As these cells are important in the regulation of Purkinje cell activity, it is likely that this results increased and aberrant inhibitory input into Purkinje cells and, thus, disrupted Purkinje cell firing and cerebellum output.
EA2: CACNA1A
Type 2 episodic ataxia (EA2) is characterized by acetazolamide-responsive attacks of ataxia with or without migraine. Patients with EA2 may also present with progressive cerebellar atrophy, nystagmus, vertigo, visual disturbances and dysarthria. These symptoms last from hours to days, in contrast with EA1, which lasts from seconds to minutes. Like EA1, attacks can be precipitated by emotional or physical stress, but also by coffee and alcohol. EA2 is caused by mutations in CACNA1A, which encodes the P/Q-type voltage-gated calcium channel CaV2.1, and is also the gene responsible for causing spinocerebellar ataxia type-6 and familial hemiplegic migraine type-1. EA2 is also referred to as episodic ataxia with nystagmus, hereditary paroxysmal cerebellopathy, familial paroxysmal ataxia and acetazolamide-responsive hereditary paroxysmal cerebellar ataxia (AHPCA). There are currently 19 mutations associated with EA2, though only 3 have been characterized electrophysiologically, table 2 and figure 2. Of these, all result in decreased current through these channels. It is assumed that the other mutations, especially the splicing and frameshift mutations, also result in a drastic decrease in CaV2.1 currents, though this may not be the case for all mutations. CACNA1A is heavily expressed in Purkinje cells of the cerebellum where it is involved in coupling action potentials with neurotransmitter release. Thus, decrease in Ca2+ entry through CaV2.1 channels is expected to result in decreased output from Purkinje cells, even though they will fire at an appropriate rate. Alternatively, some CACNA1A mutations, such as those seen in familial hemiplegic migraine type-1, result in increased Ca2+ entry and, thereby, aberrant transmitter release. This can also result in excitotoxicity, as may occur in some cases of spinocerebellar ataxia type-6.
EA3: 1q42
Episodic ataxia type-3 (EA3) is similar to EA1 but often also presents with tinnitus and vertigo. Patients typically present with bouts of ataxia lasting less than 30 minutes and occurring once or twice daily. During attacks, they also have vertigo, nausea, vomiting, tinnitus and diplopia. These attacks are sometimes accompanied by headaches and precipitated by stress, fatigue, movement and arousal after sleep. Attacks generally begin in early childhood and last throughout the patients' lifetime. Acetazolamide administration has proved successful in some patients. As EA3 is extremely rare, there is currently no known causative gene. The locus for this disorder has been mapped to the long arm of chromosome 1 (1q42).
EA4
Also known as periodic vestibulocerebellar ataxia, type-4 episodic ataxia (EA4) is an extremely rare form of episodic ataxia differentiated from other forms by onset in the third to sixth generation of life, defective smooth pursuit and gaze-evoked nystagmus. Patients also present with vertigo and ataxia. There are only two known families with EA4, both located in North Carolina. The locus for EA4 is unknown.
EA5: CACNB4
There are two known families with type-5 episodic ataxia (EA5).
These patients can present with an overlapping phenotype of ataxia and seizures similar to juvenile myoclonic epilepsy. In fact, juvenile myoclonic epilepsy and EA5 are allelic and produce proteins with similar dysfunction.
Patients with pure EA5 present with recurrent episodes of ataxia with vertigo. Between attacks they have nystagmus and dysarthria. These patients are responsive to acetazolamide.
Both juvenile myoclonic epilepsy and EA5 are a result of mutations in CACNB4, a gene that encodes the calcium channel β4 subunit. This subunit coassembles with α-subunits and produces channels that slowly inactivate after opening.
EA5 patients have a cysteine to phenylalanine mutation at position 104.
Thus results in channels with 30% greater current than wild-type.
As this subunit is expressed in the cerebellum, it is assumed that such increased current results in neuronal hyperexcitability
Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia.
EA6: SLC1A3
Type-6 episodic ataxia (EA6) is a rare form of episodic ataxia, identified initially in a 10-year-old boy who first presented with 30 minute bouts of decreased muscle tone during infancy. He required "balance therapy" as a young child to aid in walking and has a number of ataxic attacks, each separated by months to years. These attacks were precipitated by fever. He has cerebellar atrophy and subclinical seizures. During later attacks, he also presented with distortions of the left hemifield, ataxia, slurred speech, followed by headache. After enrolling in school, he developed bouts of rhythmic arm jerking with concomitant confusion, also lasting approximately 30 minutes. He also has presented, at various times, with migraines. This patient carries a proline to arginine substitution in the fifth transmembrane-spanning segment of the gene SLC1A3. This gene encodes the excitatory amino acid transporter 1 (EAAT1) protein, which is responsible for glutamate uptake. In cell culture assays, this mutation results in drastically decreased glutamate uptake in a dominant-negative manner. This is likely due to decreased synthesis or protein stability. As this protein is expressed heavily in the brainstem and cerebellum, it is likely that this mutation results in excitotoxicity and/or hyperexcitability leading to ataxia and seizures. Mutations in EAAT1 (GLAST) have subsequently been identified in a family with episodic ataxia.
Treatment
Depending on subtype, many patients find that acetazolamide therapy is useful in preventing attacks. In some cases, persistent attacks result in tendon shortening, for which surgery is required.