
 | ||
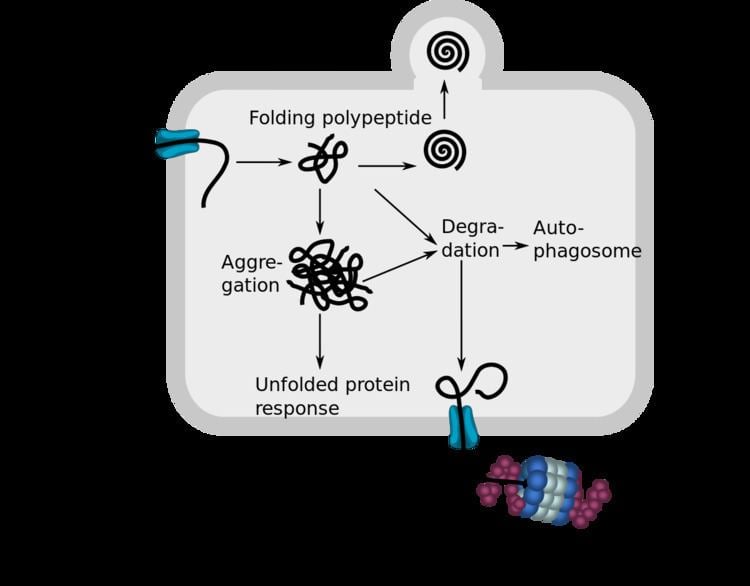
Endoplasmic-reticulum-associated protein degradation (ERAD) designates a cellular pathway which targets misfolded proteins of the endoplasmic reticulum for ubiquitination and subsequent degradation by a protein-degrading complex, called the proteasome.
Contents
Mechanism
The process of ERAD can be divided into three steps:
Recognition of misfolded or mutated proteins in the endoplasmic reticulum
The recognition of misfolded or mutated proteins depends on the detection of substructures within proteins such as exposed hydrophobic regions, unpaired cysteine residues and immature glycans.
In mammalian cells for example, there exists a mechanism called glycan processing. In this mechanism, the lectin-type chaperones calnexin/calreticulin (CNX/CRT) provide immature glycoproteins the opportunity to reach their native conformation. They can do this by way of reglucosylating these glycoproteins by an enzyme called UDP-glucose-glycoprotein glucosyltransferase. Terminally misfolded proteins, however, must be extracted from CNX/CRT. This is carried out by members of the EDEM (ER degradation-enhancing α-mannosidase-like protein) family (EDEM1-3) and ER mannosidase I. This mannosidase removes one mannose residue from the glycoprotein and the latter is recognized by EDEM. Eventually EDEM will target the misfolded glycoproteins for degradation by facilitating binding of ERAD lectins OS9 and XTP3-B.
Retro-translocation into the cytosol
Because the ubiquitin–proteasome system (UPS) is located in the cytosol, terminally misfolded proteins have to be transported from the endoplasmic reticulum back into cytoplasm. Most evidence suggest that the Hrd1 E3 ubiquitin-protein ligase can function as a retrotranslocon or dislocon to transport substrates into the cytosol. Hrd1 is not required for all ERAD events, so it is likely that other proteins contribute to this process. Further, this translocation requires a driving force that determines the direction of transport. Since polyubiquitination is essential for the export of substrates, it is widely thought that this driving force is provided by ubiquitin-binding factors. One of these ubiquitin-binding factors is the Cdc48p-Npl4p-Ufd1p complex in yeast. Humans have the homolog of Cdc48p known as valosin-containing protein (VCP/p97) with the same function as Cdc48p. VCP/p97 transports substrates from the endoplasmic reticulum to the cytoplasm with its ATPase activity.
Ubiquitin-dependent degradation by the proteasome
The ubiquitination of terminally misfolded proteins is caused by a cascade of enzymatic reactions. The first of these reactions takes place when the ubiquitin-activating enzyme E1 hydrolyses ATP and forms a high-energy thioester linkage between a cysteine residue in its active site and the C-terminus of ubiquitin. The resulting activated ubiquitin is then passed to E2, which is a ubiquitin-conjugating enzyme. Another group of enzymes, more specifically ubiquitin protein ligases called E3, bind to the misfolded protein. Next they align the protein and E2, thus facilitating the attachment of ubiquitin to lysine residues of the misfolded protein. Following successive addition of ubiquitin molecules to lysine residues of the previously attached ubiquitin, a polyubiquitin chain is formed. A polyubiquitinated protein is produced and this is recognized by specific subunits in the 19S capping complexes of the 26S proteasome. Hereafter, the polypeptide chain is fed into the central chamber of the 20S core region that contains the proteolytically active sites. Ubiquitin is cleaved before terminal digestion by deubiquitinating enzymes. This third step is very closely associated with the second one, since ubiquitination takes place during the translocation event. However, the proteasomal degradation takes place in the cytoplasm.
ERAD ubiquitination machinery
The ER membrane anchored RING finger containing ubiquitin ligases Hrd1 and Doa10 are the major mediators of substrate ubiquitination during ERAD. The tail anchored membrane protein Ubc6 as well as Ubc1 and the Cue1 dependent membrane bound Ubc7 are the ubiquitin conjugating enzymes involved in ERAD.
Checkpoints
As the variation of ERAD-substrates is enormous, several variations of the ERAD mechanism have been proposed. Indeed, it was confirmed that soluble, membrane and transmembrane proteins were recognized by different mechanisms. This led to the identification of 3 different pathways that constitute in fact 3 checkpoints.
Diseases associated with ERAD-malfunctioning
As ERAD is a central element of the secretory pathway, disorders in its activity can cause a range of human diseases. These disorders can be classified into two groups.
The first group is the result of mutations in ERAD components, which subsequently lose their function. By losing their function, these components are no longer able to stabilize aberrant proteins, so that the latter accumulate and damage the cell. An example of a disease caused by this first group of disorders is Parkinson's disease. It is caused by a mutation in the parkin gene. Parkin is a protein that functions in complex with CHIP as a ubiquitin ligase and overcomes the accumulation and aggregation of misfolded proteins.
[There are numerous theories addressing the causes of Parkinson's disease, besides the one presented here. Many of these can be found in the section of Wikipedia devoted to Parkinson's disease.]
In contrast to this first group of disorders, the second group is caused by premature degradation of secretory or membrane proteins. In this way, these proteins aren’t able to be deployed to distal compartments, as is the case in cystic fibrosis.
ERAD and HIV
As described before, the addition of polyubiquitin chains to ERAD substrates is crucial for their export. HIV uses an efficient mechanism to dislocate a single-membrane-spanning host protein, CD4, from the ER and submits it to ERAD.The Vpu protein of HIV-1 is a protein on the ER membrane and targets newly-made CD4 in the endoplasmic reticulum for degradation by cytosolic proteasomes. Vpu only utilizes part of the ERAD process to degrade CD4. CD4 is normally a stable protein and is not likely to be a target for ERAD. However, HIV produces the membrane protein Vpu that binds to CD4.The Vpu protein mainly retains the CD4 in the ER by SCFβ-TrCP-dependent ubiquitination of the CD4 cytosolic tail and transmembrane domain (TMD) interactions. The CD4 Gly415 is a contributor to CD4-Vpu interactions, several TMD-mediated mechanisms by HIV-1 Vpu are necessary to downregulate CD4 and thus promote viral pathogenesis. CD4 retained in the ER will be a target for a variant ERAD pathway rather than predominantly appearing at the plasma membrane without the presence of Vpu through the RESET pathway. Vpu mediates the CD4 retention in the ER and the addition of degradation. As Vpu is phosphorylated, it mimics substrates for the E3 complex SCFβTrCP. In cells that are infected with HIV, SCFβTrCP interacts with Vpu and ubiquitinates CD4, which is subsequently degraded by the proteasome. Vpu itself escapes from the degradation.
Questions
The big open questions related to ERAD are: