
Entrez 8050 | Ensembl ENSG00000110435 | |
 | ||
External IDs OMIM: 608769 MGI: 1351627 HomoloGene: 55757 GeneCards: PDHX | ||
E3 binding protein also known as pyruvate dehydrogenase protein X component, mitochondrial is a protein that in humans is encoded by the PDHX gene. The E3 binding protein is a component of the pyruvate dehydrogenase complex found only in eukaryotes.Defects in this gene are a cause of pyruvate dehydrogenase deficiency which results in neurological dysfunction and lactic acidosis in infancy and early childhood. This protein is also a minor antigen for antimitochondrial antibodies. These autoantibodies are present in nearly 95% of patients with the autoimmune liver disease primary biliary cirrhosis (PBC). In PBC, activated T lymphocytes attack and destroy epithelial cells in the bile duct where this protein is abnormally distributed and overexpressed. PBC eventually leads to cirrhosis and liver failure.
Contents
Structure
The mRNA encoded by the human PDHX gene is approximately 2.5 kb in length, and expressed primarily in human skeletal and cardiac muscle tissues. The gene has been localized in humans to the 11th chromosome, with the specific location being 11p1.3.
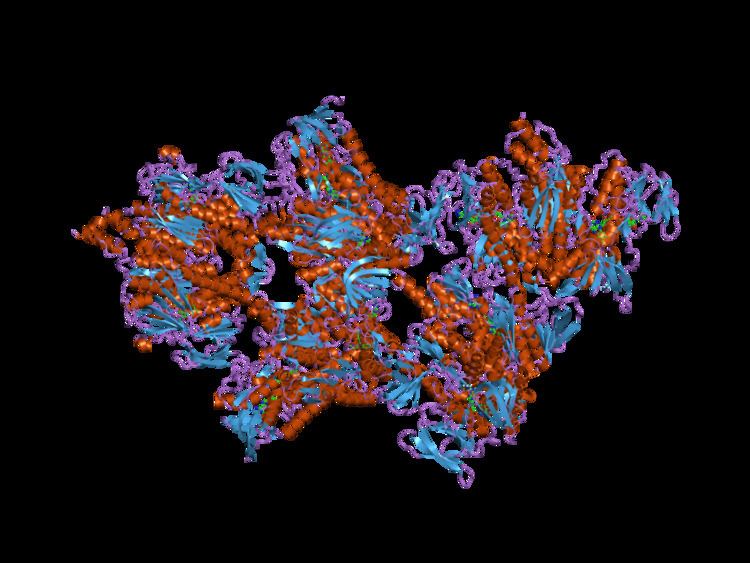
The protein encoded by the human PDHX gene, also known as E3 binding protein (E3BP), is part of the pyruvate dehydrogenase complex, a required complex for cellular respiration that catalyzes the dehydration of pyruvate to Acetyl-CoA. The entire complex is 9.5 MDa in size, and has been described as 60-meric, meaning there are over 60 components that are assembled to make the entire complex. These subunits are conserved across many species, as the function of this complex is essential for the generation of ATP for all eukaryotes. The E3 binding protein directly interacts with the dihydrolipoamide transacetylase (E2) core, anchoring it to the complex. E3BP binds the I domain of E2 by its C-terminal I' domain. The composition of E2.E3BP was thought to be 60 E2 plus approximately 12 E3BP, however, equilibrium sedimentation and small angle x-ray scattering studies showed that the E3BP/E2 binding complex has a lower mass than the E2 subunit alone. Additionally, these studies showed that E3 binds to E2.E3BP outside the central dodecahedron of the PDH complex, and that this interaction creates a lower binding affinity for the E1 subunit. Together, these data support a substitution model, in which the smaller E3BP subunits replace the E2 subunits rather than adding to the 60-mer entire complex. The specific model illustrates 12 I domains of E2 being substituted by 12 I' domains of E3BP, thereby forming 6 dimer edges that are symmetrically located in the dodecahedron structure.
E3BP similarly binds to E3, having linker regions that connect an E3-binding domain and a lipoyl domain. Crystallography of the complex has shown that, E3BD also binds to E3, though no significant conformational changes occur. In this binding, two E3 subunits come together to form the binding site. This has also shown that E3BP has residues that come into contact with the E3 component across its two-fold axis; this means that there is one binding site for this reaction on the E3 homodimer. Changing the central residues at the E3BD/E3 interface affect binding much more drastically than does changing peripheral residues. This data corroborates the theory of the existence of a “hot spot”. Specifically, there are three hydrophobic residues within the binding domain of E3BP - Pro133, Pro154, and Ile157 – that interact with the surface of both E3 polypeptide chains. This interaction is significantly stabilized by many ionic and hydrogen bonds that take place between the residues of three interacting polypeptide chains adjacent to the central hydrophobic patch. This specificity is most likely due to the lack of conformational flexibility of the binding fragment of E3BP and the complementary amino acid match with the E3 interface.
Function
The pyruvate dehydrogenase (PDH) complex is located in the mitochondrial matrix and catalyzes the conversion of pyruvate to acetyl coenzyme A. The PDH complex thereby links glycolysis to the citric acid cycle. The PDH complex contains three catalytic subunits, E1, E2, and E3, two regulatory subunits, E1 kinase and E1 phosphatase, and a non-catalytic subunit, E3 binding protein (E3BP). This gene encodes the E3 binding protein subunit; also known as component X of the pyruvate dehydrogenase complex. This protein tethers E3 dimers to the E2 core of the PDH complex.
Clinical significance
Mutations in the PDHX gene have been known to cause one form of pyruvate dehydrogenase deficiency. Pyruvate dehydrogenase deficiency is characterized by the buildup of a chemical called lactic acid in the body and a variety of neurological problems. Signs and symptoms of this condition usually first appear shortly after birth, and they can vary widely among affected individuals. The most common feature is a potentially life-threatening buildup of lactic acid (lactic acidosis), which can cause nausea, vomiting, severe breathing problems, and an abnormal heartbeat. People with pyruvate dehydrogenase deficiency usually have neurological problems as well. Most have delayed development of mental abilities and motor skills such as sitting and walking. Other neurological problems can include intellectual disability, seizures, weak muscle tone (hypotonia), poor coordination, and difficulty walking. Some affected individuals have abnormal brain structures, such as underdevelopment of the tissue connecting the left and right halves of the brain (corpus callosum), wasting away (atrophy) of the exterior part of the brain known as the cerebral cortex, or patches of damaged tissue (lesions) on some parts of the brain. Because of the severe health effects, many individuals with pyruvate dehydrogenase deficiency do not survive past childhood, although some may live into adolescence or adulthood.
While this deficiency primarily results in mutations in the E1 alpha subunit of the PDH complex, a few mutations have been identified in the PDX1 gene. Specific investigations of this gene have identified 78del85 and 965del59 mutations in a homozygous state, while some mutations could not be identified due to no PDHX mRNA being expressed in the individuals. In other cases, it has been reported that an entire exon, exon 10, was removed due to a gross deletion mutation; the mechanism for this has been theorized to be a mispairing, because there is an exact direct repeat, CCACTG, within the gene. Other large deletions (over 3900 bp) have been reported. E3BP, in coordination with the E2 subunit, has also been shown to be a secondary antigen for antimitochondrial antibodies and immune responses. The autoantibodies for this protein are present in the vast majority of patients with primary biliary cirrhosis, a chronic, progressive cholestatic liver disease that usually affects middle-aged women and eventually leads to liver failure.
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.