
 | ||
Drug development is the process of bringing a new pharmaceutical drug to the market once a lead compound has been identified through the process of drug discovery. It includes pre-clinical research on microorganisms and animals, filing for regulatory status, such as via the United States Food and Drug Administration for an investigational new drug to initiate clinical trials on humans, and may include the step of obtaining regulatory approval with a new drug application to market the drug.
Contents
New chemical entity development
Broadly, the process of drug development can be divided into pre-clinical and clinical work.
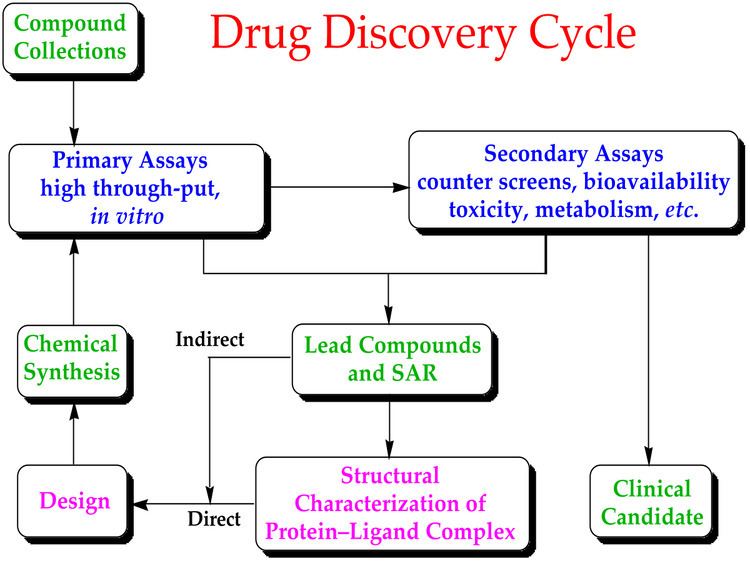
Pre-clinical
New chemical entities (NCEs, also known as new molecular entities or NMEs) are compounds that emerge from the process of drug discovery. These have promising activity against a particular biological target that is important in disease. However, little is known about the safety, toxicity, pharmacokinetics, and metabolism of this NCE in humans. It is the function of drug development to assess all of these parameters prior to human clinical trials. A further major objective of drug development is to recommend the dose and schedule for the first use in a human clinical trial ("first-in-man" [FIM] or First Human Dose [FHD]).
In addition, drug development must establish the physicochemical properties of the NCE: its chemical makeup, stability, and solubility. Manufacturers must optimize the process they use to make the chemical so they can scale up from a medicinal chemist producing milligrams, to manufacturing on the kilogram and ton scale. They further examine the product for suitability to package as capsules, tablets, aerosol, intramuscular injectable, subcutaneous injectable, or intravenous formulations. Together, these processes are known in preclinical development as chemistry, manufacturing, and control (CMC).
Many aspects of drug development focus on satisfying the regulatory requirements of drug licensing authorities. These generally constitute a number of tests designed to determine the major toxicities of a novel compound prior to first use in humans. It is a legal requirement that an assessment of major organ toxicity be performed (effects on the heart and lungs, brain, kidney, liver and digestive system), as well as effects on other parts of the body that might be affected by the drug (e.g., the skin if the new drug is to be delivered through the skin). Increasingly, these tests are made using in vitro methods (e.g., with isolated cells), but many tests can only be made by using experimental animals to demonstrate the complex interplay of metabolism and drug exposure on toxicity.
The information is gathered from this pre-clinical testing, as well as information on CMC, and submitted to regulatory authorities (in the US, to the FDA), as an Investigational New Drug application or IND. If the IND is approved, development moves to the clinical phase.
Clinical phase
Clinical trials involve three or four steps:
The process of defining characteristics of the drug does not stop once an NCE begins human clinical trials. In addition to the tests required to move a novel drug into the clinic for the first time, manufacturers must ensure that any long-term or chronic toxicities are well-defined, including effects on systems not previously monitored (fertility, reproduction, immune system, among others). They must also test the compound for its potential to cause cancer (carcinogenicity testing).
If a compound emerges from these tests with an acceptable toxicity and safety profile, and the company can further show it has the desired effect in clinical trials, then the NCE portfolio of evidence can be submitted for marketing approval in the various countries where the manufacturer plans to sell it. In the United States, this process is called a "new drug application" or NDA.
Most NCEs fail during drug development, either because they have unacceptable toxicity or because they simply do not have the intended effect on the targeted disease as shown in clinical trials.
Cost
The full cost of bringing a new drug (i.e., new chemical entity) to market – from discovery through clinical trials to approval – is complex and controversial. Typically, companies spend tens to hundreds of millions of U.S. dollars. One element of the complexity is that the much-publicized final numbers often not only include the out-of-pocket expenses for conducting a series of Phase I-III clinical trials, but also the capital costs of the long period (10 or more years) during which the company must cover out-of-pocket costs for preclinical drug discovery. Additionally, companies often do not report whether a given figure includes the capitalized cost or comprises only out-of-pocket expenses, or both.
Another element of complexity is that all estimates are based on confidential information controlled by drug companies, released by them voluntarily, leading to inability to verify costs. The numbers are controversial, as drug companies use them to justify the prices of their drugs and various advocates for lower drug prices have challenged them. The controversy is not only between "high" and "low", but also the high numbers may vary considerably for the manifold factors in drug development.
One study assessed both capitalized and out-of-pocket costs as about US$1.8 billion and $870 million, respectively.
In an analysis of the drug development costs for 98 companies over a decade, the average cost per drug developed and approved by a single-drug company was $350 million. But for companies that approved between eight and 13 drugs over 10 years, the cost per drug went as high as $5.5 billion, due mainly to geographic expansion for marketing and ongoing costs for Phase IV trials and continuous monitoring for safety.
Alternatives to conventional drug development have the objective for universities, governments and pharmaceutical industry to collaborate and optimize resources.
Valuation
The nature of a drug development project is characterised by high attrition rates, large capital expenditures, and long timelines. This makes the valuation of such projects and companies a challenging task. Not all valuation methods can cope with these particularities. The most commonly used valuation methods are risk-adjusted net present value (rNPV), decision trees, real options, or comparables.
The most important value drivers are the cost of capital or discount rate that is used, phase attributes such as duration, success rates, and costs, and the forecasted sales, including cost of goods and marketing and sales expenses. Less objective aspects like quality of the management or novelty of the technology should be reflected in the cash flows estimation.
Success rate
Candidates for a new drug to treat a disease might, theoretically, include from 5,000 to 10,000 chemical compounds. On average about 250 of these show sufficient promise for further evaluation using laboratory tests, mice and other test animals. Typically, about ten of these qualify for tests on humans. A study conducted by the Tufts Center for the Study of Drug Development covering the 1980s and 1990s found that only 21.5 percent of drugs that started Phase I trials were eventually approved for marketing. In the time period of 2006 to 2015, the success rate was 9.6%. The high failure rates associated with pharmaceutical development are referred to as the "attrition rate" problem. Careful decision making during drug development is essential to avoid costly failures. In many cases, intelligent programme and clinical trial design can prevent false negative results. Well-designed, dose-finding studies and comparisons against both a placebo and a gold-standard treatment arm play a major role in achieving reliable data.
Novel initiatives to boost development
Novel initiatives include partnering between governmental organizations and industry. The world's largest such initiative is the Innovative Medicines Initiative (IMI), and examples of major national initiatives are Top Institute Pharma in the Netherlands and Biopeople in Denmark. In 2004, the FDA created the “Critical Path Initiative” to guide the new drug development process.