
 | ||
Chromosome conformation capture techniques (often abbreviated to 3C technologies or 3C-based methods) are a set of molecular biology methods used to analyze the spatial organization of chromatin in a cell. They quantify the number of interactions between genomic loci that are nearby in 3-D space, but may be separated by many nucleotides in the linear genome. Such interactions may result from biological functions, such as promoter-enhancer interactions, or from random polymer looping, where undirected physical motion of chromatin causes loci to collide. Interaction frequencies may be analyzed directly, or they may be converted to distances and used to reconstruct 3-D structures.
Contents
- History
- Experimental methods
- 3C one vs one
- 4C one vs all
- 5C many vs many
- Hi C all vs all
- Sequence capture based methods
- Single cell methods
- ChIP loop
- ChIA PET
- Biological impact
- Data analysis
- References
The chief difference between 3C-based methods is their scope. For example, in 3C, the interactions between two specific fragments are quantified. In contrast, Hi-C quantifies interactions between all possible pairs of fragments simultaneously.
History
Historically, microscopy was the primary method of investigating nuclear organization.
In 1993, the Nuclear Ligation Assay was published, a method that could determine circularization frequencies of DNA in solution. This assay was used to show that estrogen induces an interaction between the prolactin gene promoter and a nearby enhancer.
Subsequently, some of the main ideas of the Nuclear Ligation Assay were further developed into the 3C assay, published in 2002 by Job Dekker and colleagues in the Kleckner lab at Harvard University.
Experimental methods
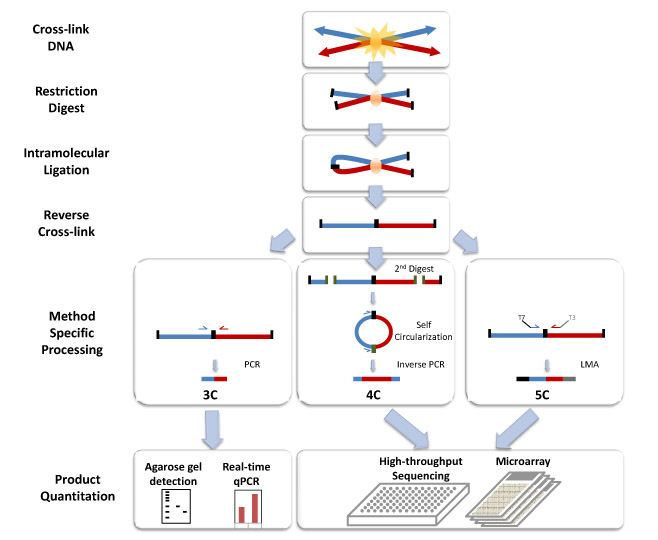
All 3C methods start with a similar set of steps, performed on a sample of cells. First, the cell genomes are cross-linked, which introduces bonds that "freeze" interactions between genomic loci. The genome is then cut into fragments. Next, random ligation is performed. This quantifies the proximity of fragments, because fragments are more likely to be ligated to nearby fragments.
Subsequently, the ligated fragments are quantified using one of a number of techniques.
3C (one-vs-one)
The chromosome conformation capture (3C) experiment quantifies interactions between a single pair of genomic loci. For example, 3C can be used to test a candidate promoter-enhancer interaction. Ligated fragments are detected using PCR with known primers.
4C (one-vs-all)
Chromosome conformation capture-on-chip (4C) captures interactions between one locus and all other genomic loci. It involves a second ligation step, to create self-circularized DNA fragments, which are used to perform inverse PCR. Inverse PCR allows the known sequence to be used to amplify the unknown sequence ligated to it. In contrast to 3C and 5C, the 4C technique does not require the prior knowledge of both interacting chromosomal regions. Results obtained using 4C are highly reproducible with most of the interactions that are detected between regions proximal to one another. On a single microarray, approximately a million interactions can be analyzed.
5C (many-vs-many)
Chromosome conformation capture carbon copy (5C) detects interactions between all restriction fragments within a given region, with this region's size typically no greater than a megabase. This is done by ligating universal primers to all fragments. However, 5C has relatively low coverage. The 5C technique overcomes the junctional problems at the intramolecular ligation step and is useful for constructing complex interactions of specific loci of interest. This approach is unsuitable for conducting genome-wide complex interactions since that will require millions of 5C primers to be used.
Hi-C (all-vs-all)
Hi-C uses high-throughput sequencing to find the nucleotide sequence of fragments. The original protocol used paired end sequencing, which retrieves a short sequence from each end of each ligated fragment. As such, for a given ligated fragment, the two sequences obtained should represent two different restriction fragments that were ligated together in the random ligation step. The pair of sequences are individually aligned to the genome, thus determining the fragments involved in that ligation event. Hence, all possible pairwise interactions between fragments are tested.
Sequence capture-based methods
A number of methods use oligonucleotide capture to enrich Hi-C libraries for specific loci of interest. These methods include Capture-C, Capture Hi-C and NG Capture-C.
Single-cell methods
Single-cell Hi-C can be used to investigate the interactions occurring in individual cells.
ChIP-loop
ChIP-loop combines 3C with ChIP-seq to detect interactions between two loci of interest mediated by a protein of interest. The ChIP-loop may be useful in identifying long-range cis-interactions and trans interaction mediated through proteins since frequent DNA collisions will not occur.
ChIA-PET
ChIA-PET combines Hi-C with ChIP-seq to detect all interactions mediated by a protein of interest.
Biological impact
3C methods have led to a number of biological insights, including the discovery of new structural features of chromosomes, the cataloguing of chromatin loops, and increased understanding of transcriptional regulation mechanisms (the disruption of which can lead to disease).
3C methods have demonstrated the importance of spatial proximity of regulatory elements to the genes that they regulate. For example, in tissues that express globin genes, the β-globin locus control region forms a loop with these genes. This loop is not found in tissues where the gene is not expressed. This technology has further aided the genetic and epigenetic study of chromosomes both in model organisms and in humans.
These methods have revealed large-scale organization of the genome into topologically associating domains (TADs), which correlate with epigenetic markers. Some TADs are transcriptionally active, while others are repressed.
Data analysis
The different 3C-style experiments produce data with very different structures and statistical properties. As such, specific analysis packages exist for each experiment type.
Hi-C data is often used to analyze genome-wide chromatin organization, such as topologically associating domains (TADs), linearly contiguous regions of the genome that are associated in 3-D space. Several algorithms have been developed to identify TADs from Hi-C data.
The 3-D organization of the genome can also be analyzed via eigendecomposition of the contact matrix. Each eigenvector corresponds to a set of loci, which are not necessarily linearly contiguous, that share structural features.
A significant confounding factor in 3C technologies is the frequent non-specific interactions between genomic loci that occur due to random polymer behavior. An interaction between two loci must be confirmed as specific through statistical significance testing.