
Specialty ophthalmology ICD-9-CM 363.55 DiseasesDB 2619 | ICD-10 H31.2 OMIM 303100 MeSH D015794 | |
 | ||
Choroideremia /kɒˌrɔɪdᵻˈriːmi.ə/ (CHM) is a rare, X-linked recessive form of hereditary retinal degeneration that affects roughly 1 in 50,000 males. The disease causes a gradual loss of vision, starting with childhood night blindness, followed by peripheral vision loss, and progressing to loss of central vision later in life. Progression continues throughout the individual's life, but both the rate of change and the degree of visual loss are variable among those affected, even within the same family.
Contents
- Disease Progression
- History
- Basic research
- Diagnosis
- Management
- Gene Therapy
- Other Potential Therapies
- Celebrities afflicted
- Organizations
- References
Choroideremia is caused by a loss-of-function mutation in the CHM gene which encodes Rab escort protein 1 (REP1), a protein involved in lipid modification of Rab proteins. While the complete mechanism of disease is not fully understood, the lack of a functional protein in the retina results in cell death and the gradual deterioration of the choroid, retinal pigment epithelium (RPE), and retinal photoreceptor cells.
As of 2017 there is no treatment for choroideremia, however retinal gene therapy clinical trials have demonstrated a possible treatment.
Disease Progression
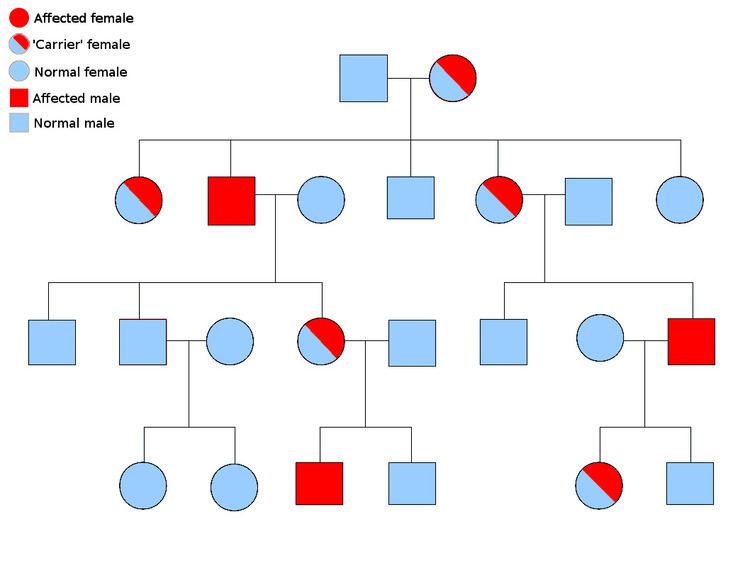
Since the CHM gene is located on the X chromosome, symptoms are seen almost exclusively in men. While there are a few exceptions, female carriers have a noticeable lack of pigmentation in the RPE but do not experience any symptoms. Female carriers have a 50% chance of having either an affected son or a carrier daughter, while a male with choroideremia will have all carrier daughters and unaffected sons. Even though the disease progression can vary significantly, there are general trends. The first symptom many individuals with choroideremia notice is a significant loss of night vision, which begins in youth. Peripheral vision loss occurs gradually, starting as a ring of vision loss, and continuing on to "tunnel vision" in adulthood. Individuals with choroideremia tend to maintain good visual acuity into their 40s, but eventual lose all sight at some point in the 50-70 age range. A study of 115 individuals with choroideremia found that 84% of patients under the age of 60 had a visual acuity of 20/40 or better, while 33% of patients over 60 years old had a visual acuity of 20/200 or worse. The most severe visual acuity impairment (only being able to count fingers or worse) did not occur until the seventh decade of life. The same study found the rate of visual acuity loss to be about 1 eye chart row per 5 years.
History
Choroideremia was first described in 1872 by an Austrian ophthalmologist, Ludwig Mauthner. Initially the condition was thought to be a developmental disorder which caused the absence of a majority of the choroid (hence the probable use of the ancient Greek suffix “eremia,” meaning barren land or desert). After several decades the stationary nature of the disease was questioned, eventually being rejected by Paymerer et al. in 1960. The CHM gene was identified and cloned in 1990 by Frans P.M. Cremers.
Basic research
In many inherited retinal diseases the protein affected by the mutation is directly involved in the light sensing function of the eye, however this is not the case in choroideremia. REP1 assists the prenylation of Rab G-proteins by binding and presenting them to the Rab geranylgeranyltransferase subunit. REP1 also escorts prenylated Rabs through the cytoplasm by binding the hydrophobic prenyl groups and carrying them to a specific destination membrane.
In healthy individuals, REP1 is found throughout all of the cells of the body, however patients with choroideremia only experience vision loss, and not broader, systemic symptoms (with the exception of a study that found crystals and fatty acid abnormalities in leukocytes). REP2, a protein that is 75% identical and 90% similar to REP1, is able to significantly compensate for the loss of REP1 outside the eye. It is thought that REP2 is not able to fully compensate for the loss of REP1 in the retina. RAB27A, a Rab that has essential functions in the retina, has been shown to be preferentially prenylated by REP1. Additionally, the Rab27a-REP1 and Rab27a-REP2 complexes have different affinities for the Rab geranylgeranyltransferase enzyme, possibly explaining REP2's inability to fully compensate for REP1 in the retina.
Diagnosis
A diagnosis of choroideremia can be made based on family history, symptoms, and the characteristic appearance of the fundus. However, choroideremia shares several clinical features with retinitis pigmentosa, a similar but broader group of retinal degenerative diseases, making a specific diagnosis difficult without genetic testing. Because of this choroideremia is often initially misdiagnosed as retinitis pigmentosa. A variety of different genetic testing techniques can be used to make a differential diagnosis.
Management
While nothing currently can be done to stop or reverse the retinal degeneration, there are steps that can be taken to slow the rate of vision loss. UV-blocking sunglasses for outdoors, appropriate dietary intake of fresh fruit and leafy green vegetables, antioxidant vitamin supplements, and regular intake of dietary omega-3 very-long-chain fatty acids are all recommended. One study found that a dietary supplement of leutein increases macular pigment levels in patients with choroideremia. Over a long period of time, these elevated levels of pigmentation could slow retinal degeneration. Additional interventions that may be needed include surgical correction of retinal detachment and cataracts, low vision services, and counseling to help cope with depression, loss of independence, and anxiety over job loss.
Gene Therapy
Gene therapy is currently not a treatment option, however human clinical trials for both choroideremia and Leber's congenital amaurosis (LCA) have produced somewhat promising results.
Clinical trials of gene therapy for patients with LCA began in 2008 at three different sites. In general, these studies found the therapy to be safe, somewhat effective, and promising as a future treatment for similar retinal diseases.
In 2011, the first gene therapy treatment for choroideremia was administered. The surgery was performed by Robert MacLaren, Professor of Ophthalmology at the University of Oxford and leader of the Clinical Ophthalmology Research Group at the Nuffield Laboratory of Ophthalmology (NLO). In the study, 2 doses of the AAV.REP1 vector were injected subretinally in 12 patients with choroideremia. There study had 2 objectives:
Despite retinal detachment caused by the injection, the study observed initial improved rod and cone function, warranting further study.
In 2016, researchers were optimistic that the positive results of 32 choroideremia patients treated over four and a half years with gene therapy in four countries could be long-lasting.
Other Potential Therapies
While choroideremia is an ideal candidate for gene therapy there are other potential therapies that could restore vision after it has been lost later in life. Foremost of these is stem cell therapy. A clinical trial published in 2014 found that a subretinal injection of human embryonic stem cells in patients with age-related macular degeneration and Stargardt disease was safe and improved vision in most patients. Out of 18 patients, vision improved in 10, improved or remained the same in 7, and decreased in 1 patient, while no improvement was seen in the untreated eyes. The study found "no evidence of adverse proliferation, rejection, or serious ocular or systemic safety issues related to the transplanted tissue." A 2015 study used CRISPR/Cas9 to repair mutations in patient-derived induced pluripotent stem cells that cause X-linked retinitis pigmentosa. This study suggests that a patient's own repaired cells could be used for therapy, reducing the risk of immune rejection and ethical issues that come with the use of embryonic stem cells.
Celebrities afflicted
A number of individuals in public roles are afflicted by choroidermia, and some have been involved in fundraising efforts for the disease. The former UK Labour Member of Parliament Siôn Simon is a known sufferer. Comic and activist E.J. Scott, partner of Daredevil actress Deborah Ann Woll, also suffers from the disease and is involved in regular fundraising efforts.
Organizations
There are a few organizations that provide support for individuals with choroideremia and raise money for research. They include: