
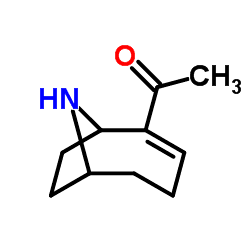
Formula C10H15NO | ||
 | ||
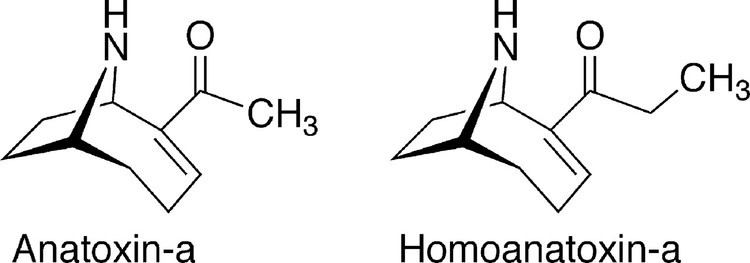
Anatoxin-a, also known as Very Fast Death Factor (VFDF), is a secondary, bicyclic amine alkaloid and cyanotoxin with acute neurotoxicity. It was first discovered in the early 1960s in Canada, and was isolated in 1972. The toxin is produced by seven different genera of cyanobacteria and has been reported in North America, Europe, Africa, Asia, and New Zealand. Symptoms of anatoxin exposure include loss of coordination, muscular fasciculations, convulsions and death by respiratory paralysis. Its mode of action is through the nicotinic acetylcholine receptor (nAchR) where it mimics the binding of the receptor's natural ligand, acetylcholine. As such, anatoxin-a has been used for medicinal purposes to investigate diseases characterized by low acetylcholine levels. Due to its high toxicity and potential presence in drinking water, anatoxin-a poses a threat to animals, including humans. While methods for detection and water treatment exist, scientists have called for more research to improve reliability and efficacy. Anatoxin-a is not to be confused with anatoxin-a(S), another potent cyanotoxin that has a similar mechanism of action to that of anatoxin-a and is produced by many of the same cyanobacteria genera, but is structurally unrelated.
Contents
- History
- Mechanism of toxicity
- Stability and Degradation
- Public Health
- Detection
- Water Treatment
- Biosynthesis
- Laboratory Uses
- Cyclic Expansion of Tropanes
- Cyclization of Cyclooctenes
- Enantioselective Enolization Strategy
- Intramolecular Cyclization of Iminium Ions
- Enyne Metathesis
- Genera of cyanobacteria that produce anatoxin a
- References

History
Anatoxin-a was first discovered by P.R Gorham in the early 1960s, after several herds of cattle died as a result of drinking water from Saskatchewan Lake in Canada, which contained toxic algal blooms. It was later isolated (1972) by J.P. Devlin from the cyanobacteria Anabaena flos aquae.
In 1977, Carmichael, Gorham, and Biggs experimented with anatoxin-a. They introduced toxic cultures of Anabaena flos aquae into the stomachs of two young male calves, and observed that muscular fasciculations and loss of coordination occurred in a matter of minutes, while death due to respiratory failure occurred anywhere between several minutes and a few hours. They also established that extensive periods of artificial respiration did not allow for detoxification to occur and natural neuromuscular functioning to resume. From these experiments, they calculated that the oral minimum lethal dose (MLD) (of the algae, not the anatoxin molecule), for calves is roughly 420 mg/kg body weight.
In the same year JP Devlin and colleagues discovered the bicyclic secondary amine structure of anatoxin-a. They also performed experiments similar to those of Carmichael et al. on mice. They found that anotoxin-a kills mice 2–5 min after intraperitoneal injection preceded by twitching, muscle spasms, paralysis and respiratory arrest. They determined the LD50 for mice to be 250 microgram/kg body weight.
Electrophysiological experiments done by Spivak et al. (1980) on frogs showed that anatoxin-a is a potent agonist of the muscle-type α12βγδ nAChR. Anatoxin-a induced depolarizing neuromuscular blockade, contracture of the frog's rectus abdominis muscle, depolarization of the frog sartorius muscle, desensitization, and alteration of the action potential. Later, Thomas et al., (1993) through his work with chicken α4β2 nAChR subunits expressed on mouse M 10 cells and chicken α7 nAChR expressed in oocytes from Xenopus laevis, showed that anatoxin-a is also a potent agonist of neuronal nAChR.
Many cases of wildlife and livestock deaths due to anatoxin-a have been reported since its discovery. Domestic dog deaths due to the cyanotoxin, as determined by analysis of stomach contents, have been observed at the lower North Island in New Zealand in 2005, in eastern France in 2003, in California of the United States in 2002 and 2006, in Scotland in 1992, and in Ireland in 1997 and 2005. In each case the dogs began showing muscle convulsions within minutes, and were dead within a matter of hours. Numerous cattle fatalities arising from the consumption of water contaminated with cyanobacteria that produce anatoxin-a have been reported in the United States, Canada, and Finland between 1980 and the present. A particularly interesting case of anatoxin-a poisoning is that of the Lesser Flamingos at Lake Bogoria in Kenya. The cyanotoxin, which was identified in the stomachs and fecal pellets of the birds, killed roughly 30,000 flamingos in the second half of 1999, and continues to cause mass fatalities annually, devastating the flamingo population. The toxin is introduced into the birds via water contaminated with cyanobacterial mat communities that arise from the hot springs in the lake bed.
Mechanism of toxicity
Anatoxin-a is an agonist of both neuronal α4β2 and α4 nicotinic acetylcholine receptors present in the CNS as well as the α12βγδ muscle-type nAchRs that are present at the neuromuscular junction. Anatoxin-a has an affinity for these receptors that is about 20 times greater than that of acetylcholine. However, the cyanotoxin has little effect on muscarinic acetylcholine receptors; it has a 100 fold lesser selectivity for these types of receptors than it has for nAchRs. Anatoxin-a also shows much less potency in the CNS than in neuromuscular junctions. In hippocampal and brain stem neurons, a 5 to 10 times greater concentration of anatoxin-a was necessary to activate nAchRs than what was required in the PNS.
In normal circumstances, acetylcholine binds to nAchRs in the post-synaptic neuronal membrane, causing a conformational change in the extracellular domain of the receptor which in turn opens the channel pore. This allows Na+ and Ca2+ ions to move into the neuron, causing cell depolarization and inducing the generation of action potentials, which allows for muscle contraction. The acetylcholine neurotransmitter then dissociates from the nAchR, where it is rapidly cleaved into acetate and choline by acetylcholinesterase.
Anatoxin-a binding to these nAchRs cause the same effects in neurons. However, anatoxin-a binding is irreversible, and the anatoxin-a nAchR complex cannot be broken down by acetylcholinesterase. Thus, the nAchR is temporarily locked open and after a period of time becomes desensitized. In this desensitized state the nAchRs no longer let cations pass through, which ultimately leads to a blockage of neuromuscular transmission.
Two enantiomers of anatoxin-a, the positive enantiomer, (+)anatoxin-a, is 150 fold more potent than the synthetic negative enantiomer, (−)anatoxin-a. This is because (+)anatoxin-a, the s-cis enone conformation, has a distance a 6.0 Å between its nitrogen and carbonyl group, which corresponds well to the 5.9 Å distance that separate the nitrogen and oxygen in acetylcholine.
Respiratory arrest, which results in a lack of an oxygen supply to the brain, is the most evident and lethal effect of anatoxin-a. Injections of mice, rats, birds, dogs, and calves with lethal doses of anatoxin-a have demonstrated that death is preceded by a sequence of muscle fasciculations, decreased movement, collapse, exaggerated abdominal breathing, cyanosis and convulsions. In mice, anatoxin-a also seriously impacted blood pressure and heart rate, and caused severe acidosis.
Stability and Degradation
Anatoxin-a is unstable in water and other natural conditions, and in the presence of UV light undergoes photodegradation, being converted to the non-toxic products dihydroanatoxin-a and epoxyanatoxin-a. The photodegradation of anatoxin-a is dependent on pH and sunlight intensity but independent of oxygen, indicating that the degradation by light is not achieved through the process of photo-oxidation.
Studies have shown that some microorganisms are capable of degrading anatoxin-a. A study done by Kiviranta and colleagues in 1991 showed that the bacterial genus Pseudomonas was capable of degrading anatoxin-a at a rate of 2–10 μg/ml per day. Later experiments done by Rapala and colleagues (1994) supported these results. They compared the effects of sterilized and non-sterilized sediments on anatoxin-a degradation over the course of 22 days, and found that after that time vials with the sterilized sediments showed similar levels of anatoxin-a as at the commencement of the experiment, while vials with non-sterilized sediment showed a 25-48% decrease.
Public Health
Despite the relatively low frequency of anatoxin-a relative to other cyanotoxins, its high toxicity (the lethal dose is not known for humans, but is estimated to be less than 5 mg for an adult male) means that it is still considered a serious threat to terrestrial and aquatic organisms, most significantly to livestock and to humans. Anatoxin-a is suspected to have been involved in the death of at least one person. The threat posed by anatoxin-a and other cyanotoxins is increasing as both fertilizer runoff, leading to eutrophication in lakes and rivers, and higher global temperatures contribute to a greater frequency and prevalence of cyanobacterial blooms.
Detection
There are two categories of anatoxin-a detection methods. Biological methods have involved administration of samples to mice and other organisms more commonly used in ecotoxicological testing, such as brine shrimp (Artemia salina), larvae of the freshwater crustacean Thamnocephalus platyurus, and various insect larvae. Problems with this methodology include an inability to determine whether it is anatoxin-a or another neurotoxin that causes the resulting deaths. Large amounts of sample material are also needed for such testing. In addition to the biological methods, scientists have used chromatography to detect anatoxin-a. This is complicated by the rapid degradation of the toxin and the lack of commercially available standards for anatoxin-a.
Water Treatment
As of now, there is no official guideline level for anatoxin-a, although scientists estimate that a level of 1 μg L−1 would be sufficiently low, the safety for drinking water being approximately 3 orders of magnitude. Likewise, there are no official guidelines regarding testing for anatoxin-a. Among methods of reducing the risk for cyanotoxins, including anatoxin-a, scientists look favorably on biological treatment methods because they do not require complicated technology, are low maintenance, and have low running costs. Few biological treatment options have been tested for anatoxin-a specifically, although a species of Pseudomonas, capable of biodegrading anatoxin-a at a rate of 2–10 μg mL−1 d−1, has been identified. Granular activated carbon (GAC) filters have also been tested as a method of biodegradation, but it is inconclusive that they were not simply absorbing the toxin. Others have called for additional studies to determine more about how to use activated carbon effectively.
More common methods of treating water, including photocatalysis UV disinfection and chlorination are not effective for targeting anatoxin-a. Other oxidants such as potassium permanganate, ozone, and the hydroxyl radical have worked in lowering levels of anatoxin-a. Optimizing the treatment process would involve the ability to remove complete cyanobacterial cells, since most of the anatoxin-a is found within the cells when the bloom is growing. Additional research needs to be done to find more reliable and efficient methods of both detection and treatment.
Biosynthesis
Anatoxin-a is synthesized in vivo in the species Anabaena flos aquae, as well as several other genera of cyanobacteria. Anatoxin-a and related chemical structures are produced using acetate and glutamate. Further enzymatic reduction of these precursors results in the formation of anatoxin-a. Homoanatoxin, a similar chemical, is produced by Oscillatoria formosa and utilizes the same precursor. However, homoanatoxin undergoes a methyl addition by S-adenosyl-L_methionine instead of an addition of electrons, resulting in a similar analogue.
Laboratory Uses
Anatoxin-a is a very powerful nicotinic acetylcholine receptor agonist and as such has been extensively studied for medicinal purposes. It is mainly used as a pharmacological probe in order to investigate diseases characterized by low acetylcholine levels, such as muscular dystrophy, Myastenia gravis, Alzheimer's disease, and Parkinson's disease. Further research on anatoxin-a and other less potent analogues are being tested as possible replacements for acetylcholine.
Cyclic Expansion of Tropanes
The first biologically occurring initial substance for tropane expansion into anatoxin-a was cocaine, which has similar stereochemistry to anatoxin-a. Cocaine is first converted into the endo isomer of cyclopropane, which is then photolytically cleaved to obtain an alpha, beta unsaturated ketone. Through the use of diethyl azodicarboxylate, the ketone is demethylated and anatoxin-a is formed. A similar, more recent synthesis pathway involves producing 2-tropinone from cocaine and treating the product with ethyl chloroformate producing a bicyclic ketone. This product is combined with trimethylsilyldiazylmethane, an organoaluminum Lewis acid and trimethylsinyl enol ether to produce tropinone. This method undergoes several more steps, producing useful intermediates as well as anatoxin-a as a final product.
Cyclization of Cyclooctenes
The first and most extensively explored approach used to synthesize anatoxin-a in vitro, cyclooctene cyclization involves 1,5-cycloocadiene as its initial source. This starting substance is reacted to form methyl amine and combined with hypobromous acid to form anatoxin-a. Another method developed in the same laboratory uses aminoalcohol in conjunction with mercuric (II) acetate and sodium borohydride. The product of this reaction was transformed into an alpha, beta ketone and oxidized by ethyl azodicarboxylate to form anatoxin-a.
Enantioselective Enolization Strategy
This method for anatoxin-a production was one of the first used that does not utilize a chimerically analogous starting substance for anatoxin formation. Instead, a racemic mixture of 3-tropinone is used with a chiral lithium amide base and additional ring expansion reactions in order to produce a ketone intermediate. Addition of an organocuprate to the ketone produces an enol triflate derivative, which is then lysed hydrogenously and treated with a deprotecting agent in order to produce anatoxin-a. Similar strategies have also been developed and utilized by other laboratories.
Intramolecular Cyclization of Iminium Ions
Iminium ion cyclization utilizes several different pathways to create anatoxin-a, but each of these produces and progresses with a pyrrolidine iminium ion. The major differences in each pathway relate to the precursors used to produce the imium ion and the total yield of anatoxin-a at the end of the process. These separate pathways include production of alkyl iminium salts, acyl iminium salts and tosyl iminium salts.
Enyne Metathesis
Enyne metathesis of anatoxin-a involves the use of a ring closing mechanism and is one of the more recent advances in anatoxin-a synthesis. In all methods involving this pathway, pyroglutamic acid is used as a starting material in conjunction with a Grubb's catalyst. Similar to iminium cyclization, the first attempted synthesis of anatoxin-a using this pathway used a 2,5-cis-pyrrolidine as an intermediate.