
 | ||
The 1000 Genomes Project, launched in January 2008, was an international research effort to establish by far the most detailed catalogue of human genetic variation. Scientists planned to sequence the genomes of at least one thousand anonymous participants from a number of different ethnic groups within the following three years, using newly developed technologies which were faster and less expensive. In 2010, the project finished its pilot phase, which was described in detail in a publication in the journal Nature. In 2012, the sequencing of 1092 genomes was announced in a Nature publication. In 2015, two papers in Nature reported results and the completion of the project and opportunities for future research. Many rare variations, restricted to closely related groups, were identified, and eight structural-variation classes were analyzed.
Contents
- Background
- Human genetic variation
- Natural selection
- Goals
- Outline
- Human genome samples
- Community meeting
- Pilot phase
- References
The project unites multidisciplinary research teams from institutes around the world, including China, Italy, Japan, Kenya, Nigeria, Peru, the United Kingdom, and the United States. Each will contribute to the enormous sequence dataset and to a refined human genome map, which will be freely accessible through public databases to the scientific community and the general public alike.
By providing an overview of all human genetic variation, not only what is already known to be biomedically relevant, the consortium will generate a valuable tool for all fields of biological science, especially in the disciplines of genetics, medicine, pharmacology, biochemistry, and bioinformatics.
Background
Since the completion of the Human Genome Project advances in human population genetics and comparative genomics have made it possible to gain increasing insight into the nature of genetic diversity. However, we are just beginning to understand how processes like the random sampling of gametes, structural variations (insertions/deletions (indels), copy number variations (CNV), retroelements), single-nucleotide polymorphisms (SNPs), and natural selection have shaped the level and pattern of variation within species and also between species.
Human genetic variation
The random sampling of gametes during sexual reproduction leads to genetic drift — a random fluctuation in the population frequency of a trait — in subsequent generations and would result in the loss of all variation in the absence of external influence. It is postulated that the rate of genetic drift is inversely proportional to population size, and that it may be accelerated in specific situations such as bottlenecks, where the population size is reduced for a certain period of time, and by the founder effect (individuals in a population tracing back to a small number of founding individuals).
Anzai et al. demonstrated that indels account for 90.4% of all observed variations in the sequence of the major histocompatibility locus (MHC) between humans and chimpanzees. After taking multiple indels into consideration, the high degree of genomic similarity between the two species (98.6% nucleotide sequence identity) drops to only 86.7%. For example, a large deletion of 95 kilobases (kb) between the loci of the human MICA and MICB genes, results in a single hybrid chimpanzee MIC gene, linking this region to a species-specific handling of several retroviral infections and the resultant susceptibility to various autoimmune diseases. The authors conclude that instead of more subtle SNPs, indels were the driving mechanism in primate speciation.
Besides mutations, SNPs and other structural variants such as copy-number variants (CNVs) are contributing to the genetic diversity in human populations. Using microarrays, almost 1,500 copy number variable regions, covering around 12% of the genome and containing hundreds of genes, disease loci, functional elements and segmental duplications, have been identified in the HapMap sample collection. Although the specific function of CNVs remains elusive, the fact that CNVs span more nucleotide content per genome than SNPs emphasizes the importance of CNVs in genetic diversity and evolution.
Investigating human genomic variations holds great potential for identifying genes that might underlie differences in disease resistance (e.g. MHC region) or drug metabolism.
Natural selection
Natural selection in the evolution of a trait can be divided into three classes. Directional or positive selection refers to a situation where a certain allele has a greater fitness than other alleles, consequently increasing its population frequency (e.g. antibiotic resistance of bacteria). In contrast, stabilizing or negative selection (also known as purifying selection) lowers the frequency or even removes alleles from a population due to disadvantages associated with it with respect to other alleles. Finally, a number of forms of balancing selection exist; those increase genetic variation within a species by being overdominant (heterozygous individuals are fitter than homozygous individuals, e.g. G6PD, a gene that is involved in both sickle cell anaemia and malaria resistance) or can vary spatially within a species that inhabits different niches, thus favouring different alleles. Some genomic differences may not affect fitness. Neutral variation, previously thought to be “junk” DNA, is unaffected by natural selection resulting in higher genetic variation at such sites when compared to sites where variation does influence fitness.
It is not fully clear how natural selection has shaped population differences; however, genetic candidate regions under selection have been identified recently. Patterns of DNA polymorphisms can be used to reliably detect signatures of selection and may help to identify genes that might underlie variation in disease resistance or drug metabolism. Barreiro et al. found evidence that negative selection has reduced population differentiation at the amino acid–altering level (particularly in disease-related genes), whereas, positive selection has ensured regional adaptation of human populations by increasing population differentiation in gene regions (mainly nonsynonymous and 5'-untranslated region variants).
It is thought that most complex and Mendelian diseases (except diseases with late onset, assuming that older individuals no longer contribute to the fitness of their offspring) will have an effect on survival and/or reproduction, thus, genetic factors underlying those diseases should be influenced by natural selection. Although, diseases that have late onset today could have been childhood diseases in the past as genes delaying disease progression could have undergone selection. Gaucher disease (mutations in the GBA gene), Crohn's disease (mutation of NOD2) and familial hypertrophic cardiomyopathy (mutations in MYH7, TNNT2, TPM1 and MYBPC3) are all examples of negative selection. These disease mutations are primarily recessive and segregate as expected at a low frequency, supporting the hypothesized negative selection. There is evidence that the genetic-basis of Type 1 Diabetes may have undergone positive selection. Few cases have been reported, where disease-causing mutations appear at the high frequencies supported by balanced selection. The most prominent example is mutations of the G6PD locus where, if homozygous G6PD enzyme deficiency and consequently sickle-cell anaemia results, but in the heterozygous state are partially protective against malaria. Other possible explanations for segregation of disease alleles at moderate or high frequencies include genetic drift and recent alterations towards positive selection due to environmental changes such as diet or genetic hitch-hiking.
Genome-wide comparative analyses of different human populations, as well as between species (e.g. human versus chimpanzee) are helping us to understand the relationship between diseases and selection and provide evidence of mutations in constrained genes being disproportionally associated with heritable disease phenotypes. Genes implicated in complex disorders tend to be under less negative selection than Mendelian disease genes or non-disease genes.
Goals
There are two kinds of genetic variants related to disease. The first are rare genetic variants that have a severe effect predominantly on simple traits (e.g. Cystic fibrosis, Huntington disease). The second, more common, genetic variants have a mild effect and are thought to be implicated in complex traits (e.g. Cognition, Diabetes, Heart Disease). Between these two types of genetic variants lies a significant gap of knowledge, which the 1000 Genomes Project is designed to address.
The primary goal of this project is to create a complete and detailed catalogue of human genetic variations, which in turn can be used for association studies relating genetic variation to disease. By doing so the consortium aims to discover >95 % of the variants (e.g. SNPs, CNVs, indels) with minor allele frequencies as low as 1% across the genome and 0.1-0.5% in gene regions, as well as to estimate the population frequencies, haplotype backgrounds and linkage disequilibrium patterns of variant alleles.
Secondary goals will include the support of better SNP and probe selection for genotyping platforms in future studies and the improvement of the human reference sequence. Furthermore, the completed database will be a useful tool for studying regions under selection, variation in multiple populations and understanding the underlying processes of mutation and recombination.
Outline
The human genome consists of approximately 3 billion DNA base pairs and is estimated to carry around 20,000 protein coding genes. In designing the study the consortium needed to address several critical issues regarding the project metrics such as technology challenges, data quality standards and sequence coverage.
Over the course of the next three years, scientists at the Sanger Institute, BGI Shenzhen and the National Human Genome Research Institute’s Large-Scale Sequencing Network are planning to sequence a minimum of 1,000 human genomes. Due to the large amount of sequence data that need to be generated and analyzed it is possible that other participants may be recruited over time.
Almost 10 billion bases will be sequenced per day over a period of the two year production phase. This equates to more than two human genomes every 24 hours; a groundbreaking capacity. Challenging the leading experts of bioinformatics and statistical genetics, the sequence dataset will comprise 6 trillion DNA bases, 60-fold more sequence data than what has been published in DNA databases over the past 25 years.
To determine the final design of the full project three pilot studies were designed and will be carried out within the first year of the project. The first pilot intends to genotype 180 people of 3 major geographic groups at low coverage (2x). For the second pilot study, the genomes of two nuclear families (both parents and an adult child) are going to be sequenced with deep coverage (20x per genome). The third pilot study involves sequencing the coding regions (exons) of 1,000 genes in 1,000 people with deep coverage (20x).
It has been estimated that the project would likely cost more than $500 million if standard DNA sequencing technologies were used. Therefore, several new technologies (e.g. Solexa, 454, SOLiD) will be applied, lowering the expected costs to between $30 million and $50 million. The major support will be provided by the Wellcome Trust Sanger Institute in Hinxton, England; the Beijing Genomics Institute, Shenzhen (BGI Shenzhen), China; and the NHGRI, part of the National Institutes of Health (NIH).
In keeping with Fort Lauderdale principles, all genome sequence data (including variant calls) is freely available as the project progresses and can be downloaded via ftp from the 1000 genomes project webpage.
Human genome samples
Based on the overall goals for the project, the samples will be chosen to provide power in populations where association studies for common diseases are being carried out. Furthermore, the samples do not need to have medical or phenotype information since the proposed catalogue will be a basic resource on human variation.
For the pilot studies human genome samples from the HapMap collection will be sequenced. It will be useful to focus on samples that have additional data available (such as ENCODE sequence, genome-wide genotypes, fosmid-end sequence, structural variation assays, and gene expression) to be able to compare the results with those from other projects.
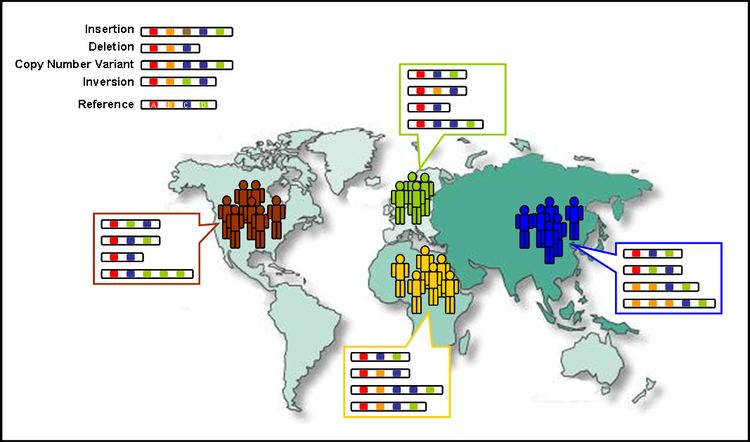
Complying with extensive ethical procedures, the 1000 Genomes Project will then use samples from volunteer donors. The following populations will be included in the study: Yoruba in Ibadan (YRI), Nigeria; Japanese in Tokyo (JPT); Chinese in Beijing (CHB); Utah residents with ancestry from northern and western Europe (CEU); Luhya in Webuye, Kenya (LWK); Maasai in Kinyawa, Kenya (MKK); Toscani in Italy (TSI); Peruvians in Lima, Peru (PEL); Gujarati Indians in Houston (GIH); Chinese in metropolitan Denver (CHD); people of Mexican ancestry in Los Angeles (MXL); and people of African ancestry in the southwestern United States (ASW).
Community meeting
Data generated by the 1000 Genomes Project is widely used by the genetics community, making the first 1000 Genomes Project one of the most cited papers in biology. To support this user community, the project held a community analysis meeting in July 2012 that included talks highlighting key project discoveries, their impact on population genetics and human disease studies, and summaries of other large scale sequencing studies.
Pilot phase
The pilot phase consisted of three projects:
It was found that on average, each person carries around 250-300 loss-of-function variants in annotated genes and 50-100 variants previously implicated in inherited disorders. Based on the two trios, it is estimated that the rate of de novo germline mutation is approximately 10−8 per base per generation.