
Specialty endocrinology ICD-9-CM 275.3 DiseasesDB 6513 | ICD-10 E83.3 OMIM 307800 | |
 | ||
eMedicine ped/1128 article/922305
MeshName = | ||
X-linked hypophosphatemia (XLH), also called X-linked dominant hypophosphatemic rickets, X-linked vitamin d-resistant rickets, is an X-linked dominant form of rickets (or osteomalacia) that differs from most cases of rickets in that ingestion of vitamin D is relatively ineffective. It can cause bone deformity including short stature and genu varum (bow leggedness). It is associated with a mutation in the PHEX gene sequence (Xp.22) and subsequent inactivity of the PHEX protein. The prevalence of the disease is 1:20000. The leg deformity can be treated with Ilizarov frames and CHAOS surgery.
Contents
Presentation
The presentation of x-linked hypophosphatemia is consistent with:
Dental Presentations:
Genetics
XLH is associated with a mutation in the PHEX gene sequence, located on the human X chromosome at location Xp22.2-p22.1. The PHEX protein regulates another protein called fibroblast growth factor 23 (produced from the FGF23 gene). Fibroblast growth factor 23 normally inhibits the kidneys' ability to reabsorb phosphate into the bloodstream. Gene mutations in PHEX prevent it from correctly regulating fibroblast growth factor 23. The resulting overactivity of FGF-23 reduces vitamin D 1α-hydroxylation and phosphate reabsorption by the kidneys, leading to hypophosphatemia and the related features of hereditary hypophosphatemic rickets. Also in XLH, where PHEX enzymatic activity is absent or reduced, osteopontin — a mineralization-inhibiting secreted substrate protein found in the extracellular matrix of bone — accumulates in bone (and teeth) to contribute to the osteomalacia (and odontomalacia) as shown in the mouse homolog (Hyp) of XLH and in XLH patients. Biochemically in blood, XLH is recognized by hypophosphatemia and an inappropriately low level of calcitriol (1,25-(OH)2 vitamin D3). Patients often have bowed legs or knock knees in which they usually cannot touch both knees and ankles together at the same time.
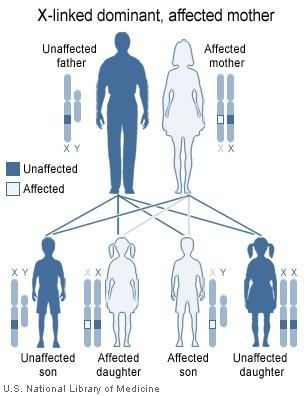
The disorder is inherited in an X-linked dominant manner. This means the defective gene responsible for the disorder (PHEX) is located on the X chromosome, and only one copy of the defective gene is sufficient to cause the disorder when inherited from a parent who has the disorder. Males are normally hemizygous for the X chromosome, having only one copy. As a result, X-linked dominant disorders usually show higher expressivity in males than females.
As the X chromosome is one of the sex chromosomes (the other being the Y chromosome), X-linked inheritance is determined by the sex of the parent carrying a specific gene and can often seem complex. This is because, typically, females have two copies of the X-chromosome and males have only one copy. The difference between dominant and recessive inheritance patterns also plays a role in determining the chances of a child inheriting an X-linked disorder from their parentage.
Treatment
Oral phosphate, 9, calcitriol, 9; in the event of severe bowing, an osteotomy may be performed to correct the leg shape.