
 | ||
Vicinal difunctionalization refers to a chemical reaction involving transformations at two adjacent centers (most commonly carbons). This transformation can be accomplished in α,β-unsaturated carbonyl compounds via the conjugate addition of a nucleophile to the β-position followed by trapping of the resulting enolate with an electrophile at the α-position. When the nucleophile is an enolate and the electrophile a proton, the reaction is called Michael addition.
Contents
Introduction
Vicinal difunctionalization reactions, most generally, lead to new bonds at two adjacent carbon atoms. Often this takes place in a stereocontrolled fashion, particularly if both bonds are formed simultaneously, as in the Diels-Alder reaction. Activated double bonds represent a useful handle for vicinal difunctionalization because they can act as both nucleophiles and electrophiles—one carbon is necessarily electron poor, and the other electron rich. In the presence of a nucleophile and an electrophile, then, the two carbons of a double bond can act as a "relay," mediating electron flow from the nucleophile to the electrophile with the formation of two, rather than the usual one, chemical bonds.
(1)
Most often, the nucleophile employed in this context is an organometallic compound and the electrophile is an alkyl halide.
Prevailing mechanism
The mechanism proceeds in two stages: β-nucleophilic addition to the unsaturated carbonyl compound, followed by electrophilic substitution at the α-carbon of the resulting enolate.
When the nucleophile is an organometallic reagent, the mechanisms of the first step can vary. Whether reactions take place by ionic or radical mechanisms is unclear in some cases. Research has shown that the second step may even proceed via single-electron transfers when the reduction potential of the electrophile is low. A general scheme involving ionic intermediates is shown below.
(2)
Lithium organocuprates undergo oxidative addition to enones to give, after reductive elimination of an organocopper(III) species, β-substituted lithium enolates.
In any case, the second step is well described in all cases as the reaction of an enolate with an electrophile. The two steps may be carried out as distinct experimental operations if the initially formed enolate is protected after β-addition. If the two steps are not distinct, however, the counterion of the enolate is determined by the counterion of the nucleophilic starting material and can influence the reactivity of the enolate profoundly.
Stereochemistry
Steric approach control is common in conjugate addition reactions. Thus, in cyclic substrates, a trans relationship between substituents on the α- and β-carbons is common. The configuration at the α-position is less predictable, especially in cases when epimerization can occur. On the basis of steric approach control, the new α-substituent is predicted to be trans to the new β-substituent, and this is observed in a number of cases.
(3)
Nucleophiles and electrophiles
Organocopper reagents are the most common nucleophiles for the β-addition step. These reagents can be generated catalytically in the presence of Grignard reagents using either copper(I) or copper(II) salts.
(4)
Copper reagents can also be used stoichiometrically, and among these, organocuprates are the most common (they are more reactive than the corresponding neutral organocopper(I) compounds). The cuprate counterion may affect the addition and subsequent enolate reaction in subtle ways. Additions involving higher-order cuprates must be quenched with a silyl halide before alkylation. (5)
When unsymmetrical cuprates are employed, the group whose carbon-copper bond contains less s character is almost always transferred to the β-position. A few exceptions exist, however. In the example below, conducting the reaction in THF led to transfer of the vinyl moiety, while other solvents promoted methyl transfer.
(6)
Enolates can also be used as nucleophiles for vicinal difunctionalization reactions. To prevent simple Michael addition (which culminates in protonation of the enolate intermediate), trapping by the electrophile must be intramolecular.
(7)
Considerations of the electrophile should take into account the nature of the conjugate enolate generated after the first step. Relatively reactive alkylating agents should be used, especially in cases involving the addition of cuprates (enolates resulting from the addition of cuprates are often unreactive). Oxophilic electrophiles should be avoided, if C-alkylation is desired. Electrophiles should also lack hydrogens acidic enough to be deprotonated by an enolate.
α,β-Unsaturated carbonyl compounds
Cyclic α,β-unsaturated ketones are the most commonly employed substrates for vicinal difunctionalization. They tend to be more reactive than acyclic analogues and undergo less direct addition than aldehydes. Amides and esters can be used to encourage conjugate addition in cases when direct addition may be competitive (as in the addition of organolithium compounds).
(8)
Because the addition step is highly sensitive to steric effects, β-substituents are likely to slow the reaction. Acetylenic and allenic substrates react to give products with some retained unsaturation.
(9)
Synthetic applications
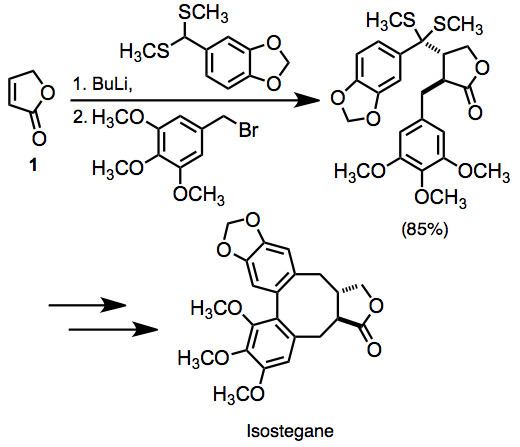
A large number of examples of vicinal difunctionalization of unsaturated carbonyl compounds exist in the literature. In one example, the difunctionalization of unsaturated lactone 1 was employed en route to isostegane. This transformation was accomplished in one pot.
(10)
Because the reaction creates two new bonds with a moderately high degree of stereocontrol, it represents a highly convergent synthetic method.
Typical conditions
Organometallic nucleophiles used for conjugate additions are most often prepared in situ. The use of anhydrous equipment and inert atmosphere is necessary. Because these factors are sometimes difficult to control and the strength of freshly prepared reagents can vary substantially, titration methods are necessary to verify the purity of reagents. A number of efficient titration methodologies exist.
Usually, vicinal difunctionalizations are carried out in one pot, without the intermediacy of a neutral protected enolate. However, in specific cases it may be necessary to protect the intermediate of β-addition. Before reaching this point, however, solvent and nucleophile screens, order of addition adjustments, and counterion adjustments can be made to optimize the one-pot process for a particular combination of carbonyl compound, nucleophile, and alkylating (or acylating) agent. Solvent adjustments between the two steps are common; if one solvent is used, tetrahydrofuran is the solvent of choice. Polar aprotic solvents should be avoided for the conjugate addition step. Concerning temperature, conjugate additions are usually carried out at low temperatures (-78 °C), while alkylations are carried out at slightly higher temperatures (0 to -30 °C). Less reactive alkylating agents may require room temperature.
Example procedure
(11)
To 6.25 g (50 mmol) of 4,4-dimethyl-2-cyclohexen-1-one and 0.5 g (5.6 mmol) of cuprous cyanide in 400 mL of diethyl ether at –23° under argon was added 100 mL (~0.75 M in diethyl ether) of 5-trimethylsilyl-4-pentynylmagnesium iodide during 4 hours. Methyl chloroformate (8 mL, 100 mmol) was added and stirring continued for 1 hour at –23° and 0.5 hour at room temperature. Hydrochloric acid (100 mL, 2.0 M) then was added and the organic phase separated and dried with magnesium sulfate. The solvent was removed and the residue chromatographed on silica gel using 5% diethyl ether–petroleum ether to give methyl 3,3-dimethyl-6-oxo-2-[5-(trimethylsilyl)-4-pentynyl]cyclohexanecarboxylate, 9.66 g (60%). IR 2000, 2140, 1755, 1715, 1660, 1615, 1440, 1280, 1250, 1225, 1205, and 845 cm–1; 1H NMR ( CDCl3) δ 0.13 (s, 9H), 0.93 (s, 3H), 1.02 (s, 3H), 1.2–2.3 (m, 11H), 3.74 (s, 3H). Anal. Calc. for C18H30O3Si: C, 67.05; H, 9.4. Found: C, 67.1; H, 9.65.