
Entrez 1200 | Ensembl ENSG00000166340 | |
 | ||
Aliases TPP1, CLN2, LPIC, SCAR7, TPP-1, GIG1, Tripeptidyl peptidase I, tripeptidyl peptidase 1 External IDs MGI: 1336194 HomoloGene: 335 GeneCards: TPP1 | ||
Tripeptidyl-peptidase 1, also known as Lysosomal pepstatin-insensitive protease, is an enzyme that in humans is encoded by the TPP1 gene. TPP1 should not be confused with the TPP1 shelterin protein which protects telomeres and is encoded by the ACD gene. Mutations in the TPP1 gene leads to late infantile neuronal ceroid lipofuscinosis.
Contents
Gene
The human gene TPP1 encodes a member of the sedolisin family of serine proteases. The human gene has 13 exons and locates at the chromosome band 11p15.
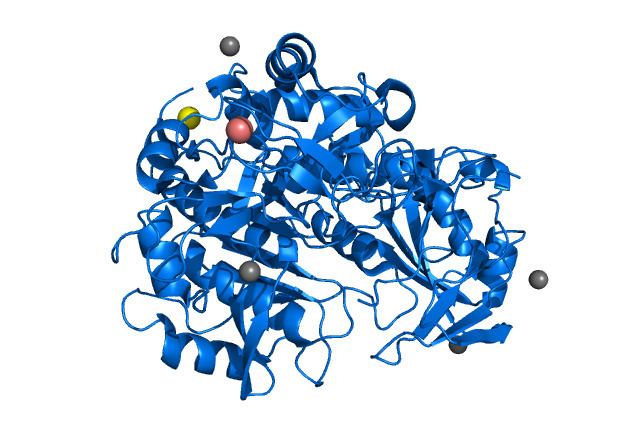
Protein
The human TPP1 is 61kDa in size and composed of 563 amino acids. An isoform of 34.5kDa and 320 amino acids is generated by alternative splicing and a peptide fragment of 1-243 amino acid is missing. TPP1 contains a globular structure with a subtilisin-like fold, a Ser475-Glu272-Asp360 catalytic triad. It also contains an octahedrally coordinated Ca2+-binding site that are characteristic features of the S53 sedolisin family of peptidases. Unlike other S53 peptidases, it has steric constraints on the P4 substrate pocket, which might contribute to its preferential cleavage of tripeptides from the unsubstituted N-terminus of proteins. Two alternative conformations of the catalytic Asp276 are associated with the activation status of TPP1.
Function
High expression of TPP1 is found in bone marrow, placenta, lung, pineal and lymphocytes. The protease functions in the lysosome to cleave N-terminal tripeptides from substrates and has weaker endopeptidase activity. It is synthesized as a catalytically inactive enzyme which is activated and autoproteolyzed upon acidification.
Clinical significance
The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited neurodegenerative disorders with pathological phenotypes that auto fluorescent lipopigments present in neurons and other cell types. Over the past two decades, accumulating evidences indicates that NCLs are caused by mutations in eight different genes, including genes encoding several soluble proteins (cathepsin D, PPT1, and TPP1). Mutations of gene TPP1 result in late-infantile neuronal ceroid lipofuscinosis which is associated with the failure to degrade specific neuropeptides and a subunit of ATP synthase in the lysosome. Mutations in the TPP1 gene lead to late infantile neuronal ceroid lipofuscinosis, a fatal neurodegenerative disease of childhood. It has been demonstrated that a single injection of intravitreal implantation of autologous bone marrow derived stem cells transduced with a TPP1 expression construct at an early stage in the disease progression could substantially inhibite the development of disease-related retinal function deficits and structural changes. This result implies that ex vivo gene therapy using autologous stem cells may be an effective means of achieving sustained delivery of therapeutic compounds to tissues such as the retina for which systemic administration would be ineffective.