
 | ||
Anticancer chemotherapy topoisomerase inhibitors part 1
Topoisomerase inhibitors are chemical compounds that block the action of topoisomerase (topoisomerase I and II), which are enzymes that control the changes in DNA structure by catalyzing the breaking and rejoining of the phosphodiester backbone of DNA strands during the normal cell cycle.
Contents
- Anticancer chemotherapy topoisomerase inhibitors part 1
- Anticancer chemotherapy topoisomerase inhibitors part 2
- Classification
- Type I topoisomerase inhibitors
- Type II topoisomerase inhibitors
- Topo II poisons
- Topo II inhibitors
- Synthetic lethality with deficient WRN expression
- References
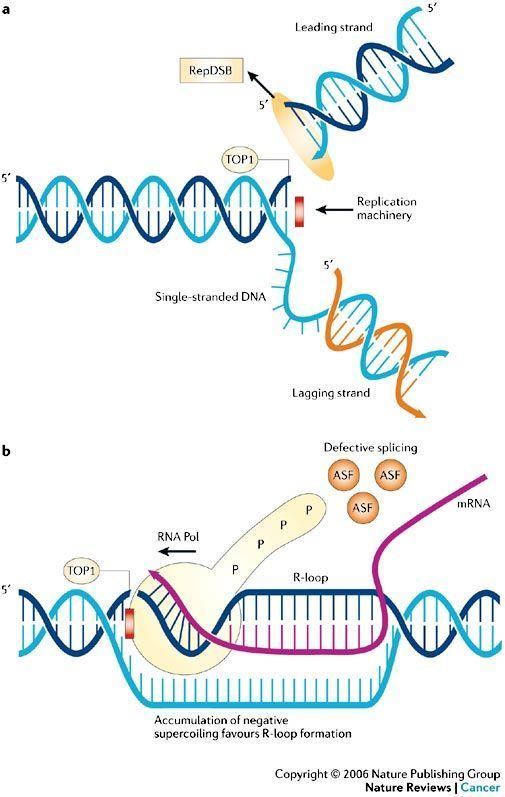
In recent years, topoisomerases have become popular targets for cancer chemotherapy treatments. It is thought that topoisomerase inhibitors block the ligation step of the cell cycle, generating single and double stranded breaks that harm the integrity of the genome. Introduction of these breaks subsequently leads to apoptosis and cell death.
Topoisomerase inhibitors can also function as antibacterial agents. Quinolones (including nalidixic acid and ciprofloxacin) have this function. Quinolones bind to these enzymes and prevent them from decatenation replicating DNA.
Anticancer chemotherapy topoisomerase inhibitors part 2
Classification
Topoisomerase inhibitors are often divided according to which type of enzyme it inhibits.
Numerous plant derived natural phenols (ex. EGCG, genistein, quercetin, resveratrol) possess strong topoisomerase inhibitory properties affecting both types of enzymes. They may express function of phytoalexins - compounds produced by plants to combat vermin and pests.
Use of topoisomerase inhibitors for antineoplastic treatments may lead to secondary neoplasms because of DNA damaging properties of the compounds. Also plant derived polyphenols shows signs of carcinogenity, especially in fetuses and neonates who do not detoxify the compounds sufficiently. An association between high intake of tea (containing polyphenols) during pregnancy and elevated risk of childhood malignant central nervous system (CNS) tumours has been found.
Type I topoisomerase inhibitors
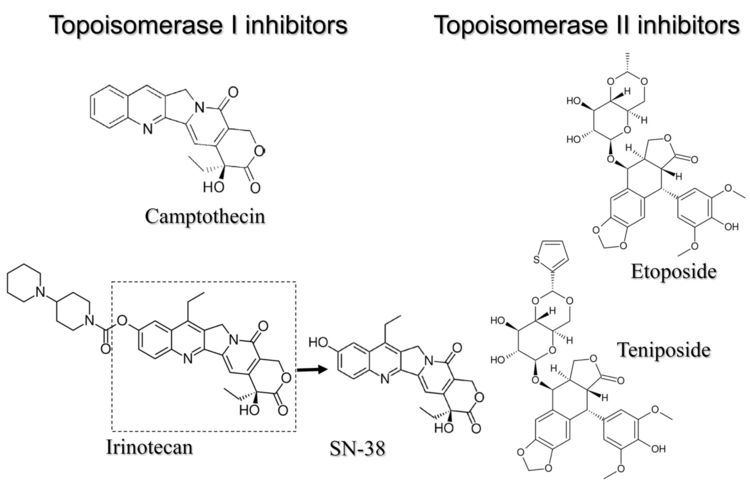
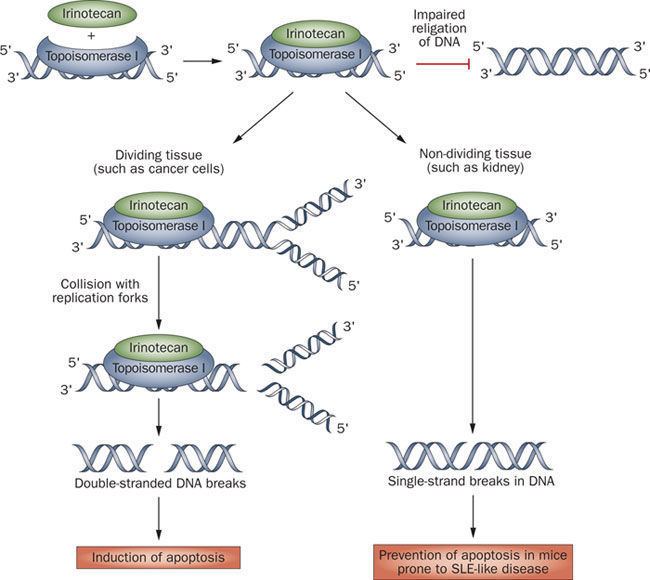
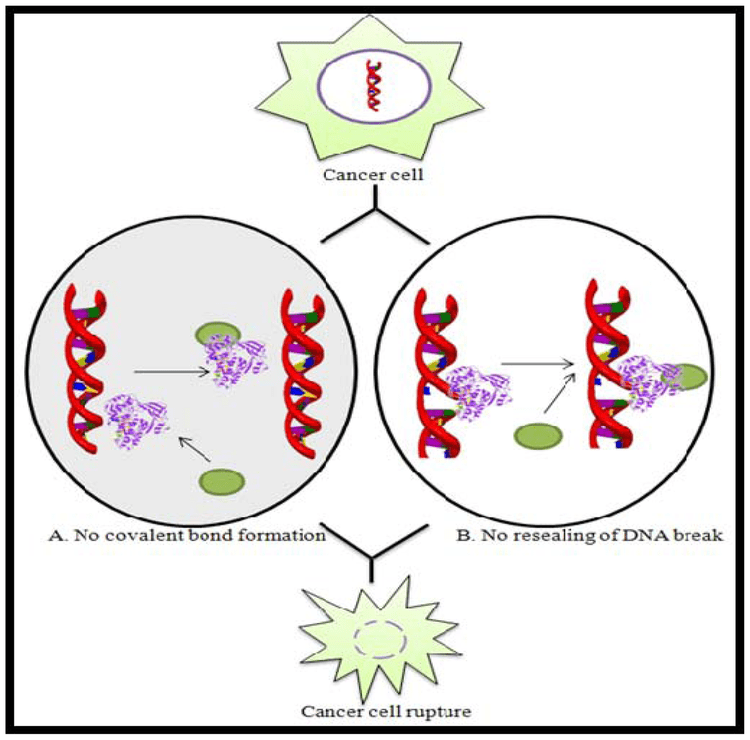
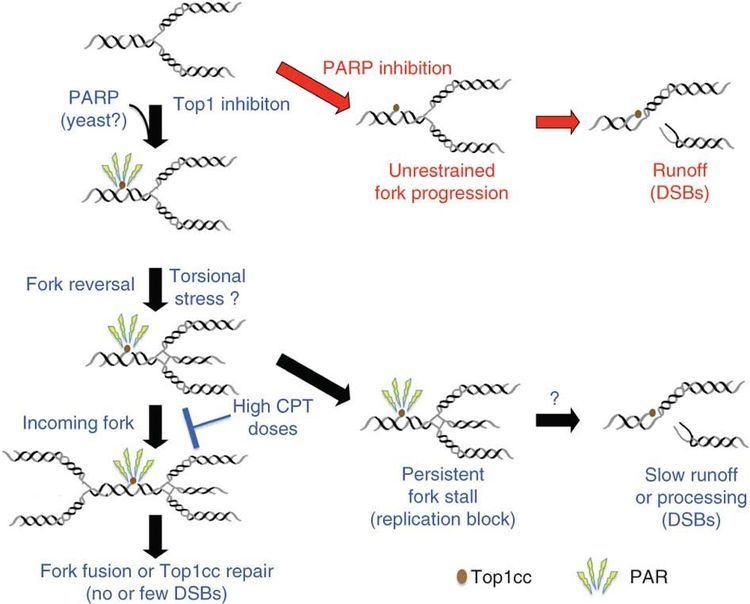
Human DNA topoisomerase I (Top1) is an essential enzyme that relaxes DNA supercoiling during replication and transcription. Top1 generates DNA single-strand breaks that allow rotation of the cleaved strand around the double helix axis. Top1 also religates the cleaved strand to reestablish intact duplex DNA. The Top1-DNA intermediates, known as cleavage complexes, are transient and at low levels under normal circumstances. However, treatment with Top1 inhibitors, such as the camptothecins, stabilize the cleavable complexes, prevent DNA religation and induce lethal DNA strand breaks. Cancer cells are selectively sensitive to the generation of these DNA lesions.
Top1 is a validated target for the treatment of human cancers. Camptothecins are among the most effective anticancer agents recently introduced into clinical practice. In this regard, the camptothecin derivative topotecan (Hycamtin) is approved by the U.S. FDA for the treatment of ovarian and lung cancer. Another camptothecin derivative irinotecan (CPT11) is approved for the treatment of colon cancer.
There are, however, certain clinical limitations of the camptothecin derivatives. These include: 1) spontaneous inactivation to a lactone form in blood, 2) rapid reversal of the trapped cleavable complex after drug removal, requiring prolonged infusions, 3) resistance of cancer cells overexpressing membrane transporters, and 4) dose-limiting side effects of diarrhea and neutropenia.
To circumvent these limitations, Dr. Mark Cushman at Purdue University and Dr. Yves Pommier at the National Cancer Institute developed the non-camptothecin family of indenoisoquinoline inhibitors of Top1. In contrast to the camptothecins, the indenoisoquinolines are: 1) chemically stable in blood, 2) inhibitors of Top1 cleavable complexes at distinct sites, 3) not substrates of membrane transporters, and 4) more effective as anti-tumor agents in animal models. The preclinical and IND package filed with the US Food and Drug Administration along with complete GMP production supporting the lead molecule are components of the published and non-published information covered by the license agreement with Purdue Research Foundation the National Cancer Center and Linus Oncology, Inc.
Linus Oncology has licensed the intellectual property that covers the development of these and related indenoisoquinoline derivatives. Phase I Study in Adults With Relapsed Solid Tumors and Lymphomas is ongoing (2012).
Indenoisoquinolines (green) form a ternary complexes of Top1 (brown) and DNA (blue) (Pommier et al.) and act as interfacial inhibitors.
There are several advantages of these novel non-camptothecin Top1 inhibitors as compared to the FDA-approved camptothecin analogs:
Based on these highly favorable characteristics, two indenoisoquinoline derivatives (from a series of > 400 molecules), indotecan (LMP400; NSC 743400) and indimitecan (LMP776; NSC 725776) are presently under evaluation in a Phase I clinical trial being conducted at the National Cancer Institute for patients with relapsed solid tumors and lymphomas. http://clinicaltrials.gov/ct2/show/NCT01051635
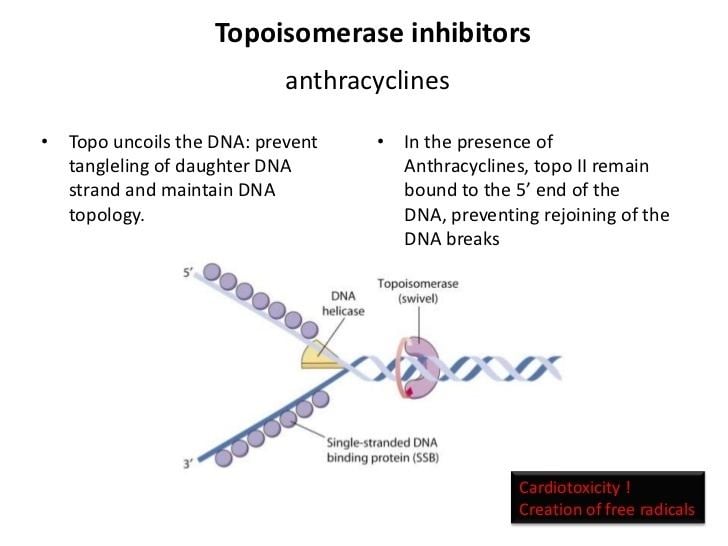
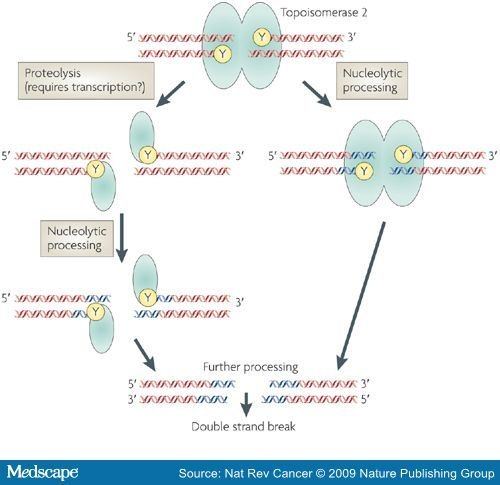
Type II topoisomerase inhibitors
These inhibitors are split into two main classes: topoisomerase poisons, which target the topoisomerase-DNA complex, and topoisomerase inhibitors, which disrupt catalytic turnover.
Topo II poisons
Examples of topoisomerase poisons include the following:
Some of these poisons encourage the forward cleavage reaction (fluoroquinolones), while other poisons prevent the re-ligation of DNA (etoposide and teniposide).
Interestingly, poisons of type IIA topoisomerases can target prokaryotic and eukaryotic enzymes preferentially, making them attractive drug candidates. Ciprofloxacin targets prokaryotes in excess of a thousandfold more than it targets eukaryotic topo IIs. The mechanism for this specificity is unknown, but drug-resistant mutants cluster in regions around the active site.
Topo II inhibitors
These inhibitors target the N-terminal ATPase domain of topo II and prevent topo II from turning over.
Examples of topoisomerase inhibitors include :
Synthetic lethality with deficient WRN expression
Synthetic lethality arises when a combination of deficiencies in the expression of two or more genes leads to cell death, whereas a deficiency in expression of only one of these genes does not. The deficiencies can arise through mutations, epigenetic alterations or inhibitors of the genes. Synthetic lethality with the topoisomerase inhibitor irinotecan appears to occur when given to cancer patients with deficient expression of the DNA repair gene WRN.
The analysis of 630 human primary tumors in 11 tissues shows that hypermethylation of the WRN CpG island promoter (with loss of expression of WRN protein) is a common event in tumorigenesis. WRN is repressed in about 38% of colorectal cancers and non-small-cell lung carcinomas and in about 20% or so of stomach cancers, prostate cancers, breast cancers, non-Hodgkin lymphomas and chondrosarcomas, plus at significant levels in the other cancers evaluated. The WRN protein helicase is important in homologous recombinational DNA repair and also has roles in non-homologous end joining DNA repair and base excision DNA repair.
A 2006 retrospective study, with long clinical follow-up, was made of colon cancer patients treated with the topoisomerase inhibitor irinotecan. In this study, 45 patients had hypermethylated WRN gene promoters and 43 patients had unmethylated WRN promoters. Irinotecan was more strongly beneficial for patients with hypermethylated WRN promoters (39.4 months survival) than for those with unmethylated WRN promoters (20.7 months survival). Thus, a topoisomerase inhibitor appeared to be especially synthetically lethal with deficient WRN expression. Further evaluations have also indicated synthetic lethality of deficient expression of WRN and topoisomerase inhibitors.