
Specialty endocrinology ICD-9-CM 272.5 DiseasesDB 12901 | ICD-10 E78.6 OMIM 205400 MeSH D013631 | |
 | ||
Tangier disease (also known as Familial alpha-lipoprotein deficiency) or Hypoalphalipoproteinemia is a rare inherited disorder characterized by a severe reduction in the amount of high density lipoprotein (HDL), often referred to as "good cholesterol," in the bloodstream.
Contents
Signs/symptoms
Individuals that are homozygotes for Tangier's disease develop various cholesterol ester depositions. These are especially visible in the tonsils, as they may appear yellow/orange. The cholesterol esters may also be found in lymph nodes, bone marrow, the liver and spleen.
Due to the cholesterol ester depositions the tonsils may be enlarged. Hepatosplenomegaly (enlarged liver and spleen) is common.
Neuropathy and cardiovascular disease are the most devastating developments caused by Tangier's disease.
Genetics
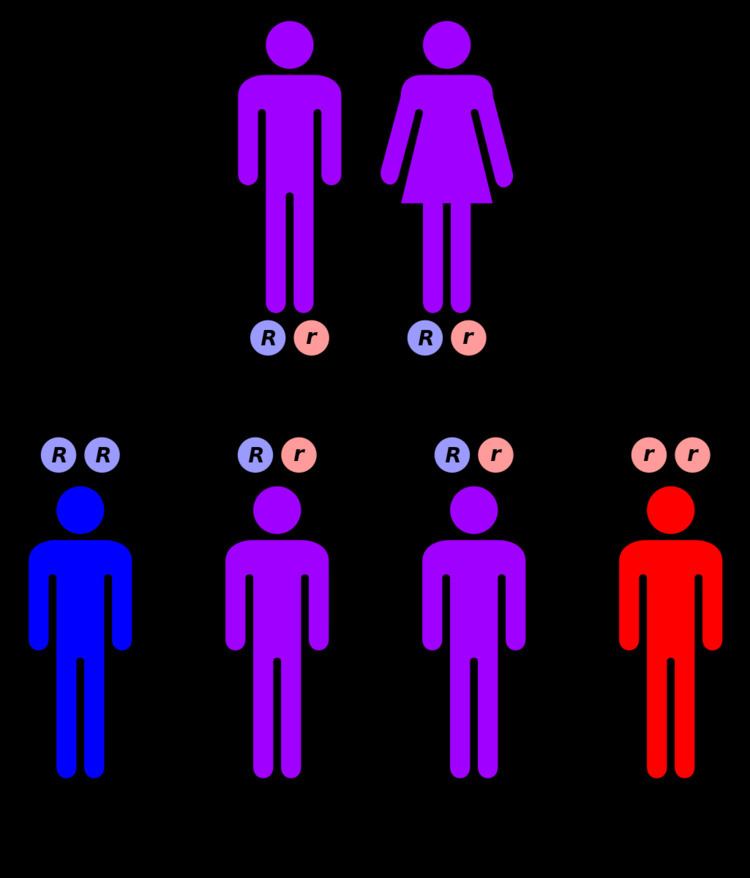
Mutations to chromosome 9q31 lead to a defective ABCA1 transporter. These mutations prevent the ABCA1 protein from effectively transporting cholesterol and phospholipids out of cells for pickup by ApoA1 in the bloodstream. This inability to transport cholesterol out of cells leads to a deficiency of high-density lipoproteins in the circulation, which is a risk factor for coronary artery disease. Additionally, the buildup of cholesterol in cells can be toxic, causing cell death or impaired function. These combined factors lead to the signs and symptoms of Tangier disease.
This condition is inherited in an autosomal recessive pattern, meaning that for the phenotype to appear, two copies of the gene must be present in the genotype. Most often, the parents of an individual with an autosomal recessive disorder are carriers of one copy of the altered gene but do not show signs and symptoms of the disorder.
Diagnosis
High-density lipoproteins are created when a protein in the bloodstream, Apolipoprotein A1 (apoA1), combines with cholesterol and phospholipids. The cholesterol and phospholipids used to form HDL originate from inside cells but are transported out of the cell into the blood via the ABCA1 transporter. People with Tangier disease have defective ABCA1 transporters resulting in a greatly reduced ability to transport cholesterol out of their cells, which leads to an accumulation of cholesterol and phospholipids in many body tissues, which can cause them to increase in size. Reduced blood levels of high-density lipoproteins is sometimes described as hypoalphalipoproteinemia.
People affected by this condition also have slightly elevated amounts of fat in the blood (mild hypertriglyceridemia) and disturbances in nerve function (neuropathy). The tonsils are visibly affected by this disorder; they frequently appear orange or yellow and are extremely enlarged. Affected people often develop premature atherosclerosis, which is characterized by fatty deposits and scar-like tissue lining the arteries. Other signs of this condition may include an enlarged spleen (splenomegaly), an enlarged liver (hepatomegaly), clouding of the cornea, and early-onset cardiovascular disease.
Tangier disease is a rare disorder with approximately 50 cases identified worldwide. This disorder was originally discovered on Tangier Island off the coast of Virginia, but has now been identified in people from many different countries.
Treatment
There are drugs that can increase serum HDL such as niacin or gemfibrozil. While these drugs are useful for patients with hyperlipidemia, Tangier's disease patients do not benefit from these pharmaceutical interventions.
Therefore the only current treatment modality for Tangier's disease is diet modification. A low-fat diet can reduce some of the symptoms, especially those involving neuropathies.
History
In 1959, a five-year-old patient named Teddy Laird from Tangier Island, Virginia, presented with strikingly large and yellow-orange tonsils which were removed by armed forces physicians. After initial diagnosis with Niemann-Pick he was transferred to Dr. Louis Avioli at the National Cancer Institute. Donald Fredrickson, then head of the Molecular Disease Branch, became aware of the case and had a hunch that the original diagnosis was incorrect. In 1960 he traveled with Dr. Avioli to Tangier Island for further investigation. After finding the same symptom in Teddy's sister and investigation revealing an extremely high number of foam cells in not only the tonsils but a wide range of tissues including the bone marrow and spleen, a second trip to the island was made and the discovery was made of very low HDL cholesterol in both the sister and parents of Teddy, evidence for a genetic basis of the disease.