
ICD-10 C49.9 OMIM 300813 MeSH D013584 | ICD-O M9040/3-9043/3 DiseasesDB 34577 | |
 | ||
A synovial sarcoma (also known as: malignant synovioma) is a rare form of cancer which occurs primarily in the extremities of the arms or legs, often in close proximity to joint capsules and tendon sheaths. As one of the soft tissue sarcomas, it is one of the rarest forms of soft tissue cancer.
Contents
The name "synovial sarcoma" was coined early in the 20th century, as some researchers thought that the microscopic similarity of some tumors to synovium, and its propensity to arise adjacent to joints, indicated a synovial origin; however, the actual cells from which the tumor develops are unknown and not necessarily synovial.
Primary synovial sarcomas are most common in the soft tissue near the large joints of the arm and leg but have been documented in most human tissues and organs, including the brain, prostate, and heart.
Synovial sarcoma occurs most commonly in the young, representing about 8% of all soft tissue sarcomas but about 15–20% of cases occur in adolescents and young adults. The peak of incidence is in the third decade of life, with males being affected more often than females (ratio around 1.2:1).
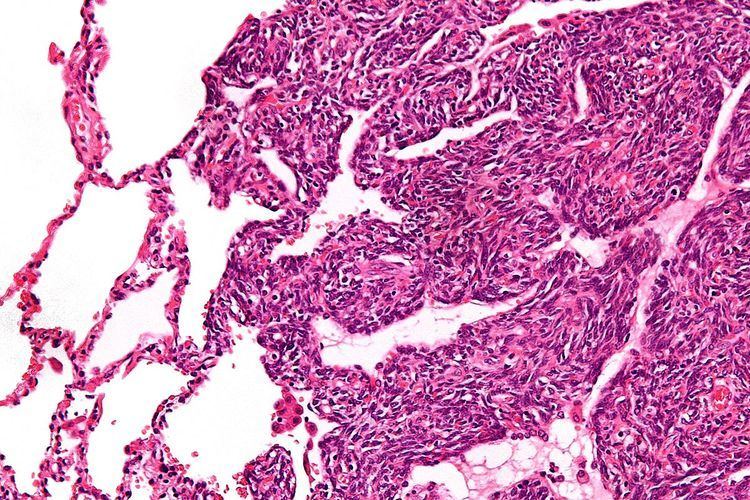
Histopathology
Two cell types can be seen microscopically in synovial sarcoma. One fibrous type, known as a spindle or sarcomatous cell, is relatively small and uniform, and found in sheets. The other is epithelial in appearance. Classical synovial sarcoma has a biphasic appearance with both types present. Synovial sarcoma can also appear to be poorly differentiated or to be monophasic fibrous, consisting only of sheets of spindle cells. Some authorities state that, extremely rarely, there can be a monophasic epithelial form which causes difficulty in differential diagnosis. Depending on the site, there is similarity to biphenotypic sinonasal sarcoma, although the genetic findings are distinctive.
Like other soft tissue sarcomas, there is no universal grading system for reporting histopathology results. In Europe, the Trojani or French system is gaining in popularity while the NCI grading system is more common in the United States. The Trojani system scores the sample, depending on tumour differentiation, mitotic index, and tumour necrosis, between 0 and 6 and then converts this into a grade of between 1 and 3, with 1 representing a less aggressive tumour. The NCI system is also a three-grade one, but takes a number of other factors into account.
Molecular biology
Most, and perhaps all, cases of synovial sarcoma are associated with a reciprocal translocation t(x;18)(p11.2;q11.2). There is some debate about whether the molecular observation itself is definitional of synovial sarcoma.
The diagnosis of synovial sarcoma is typically made based on histology and is confirmed by the presence of t(X;18). This translocation event between the SS18 gene on chromosome 18 and one of 3 SSX genes (SSX1, SSX2 and SSX4) on chromosome X causes the presence of a SS18-SSX fusion gene. The resulting fusion protein brings together the transcriptional activating domain of SS18 and the transcriptional repressor domains of SSX. It also incorporates into the SWI/SNF chromatin remodeling complex, a well known tumor suppressor. SS18-SSX is thought to underlie synovial sarcoma pathogenesis through dysregulation of gene expression.
There is some association between the SS18-SSX1 or SS18-SSX2 fusion type and both tumour morphology and five-year survival.
Symptoms
Synovial sarcoma usually presents with an otherwise asymptomatic swelling or mass, although general symptoms related to malignancies can be reported such as fatigue.
Treatment
Treatment is usually multimodal, involving surgery, chemotherapy and radiotherapy: