
Specialty endocrinology ICD-9-CM 277.5 MedlinePlus 001210 | ICD-10 E76.2 DiseasesDB 29177 | |
 | ||
OMIM 252900 252920 252940 252930 | ||
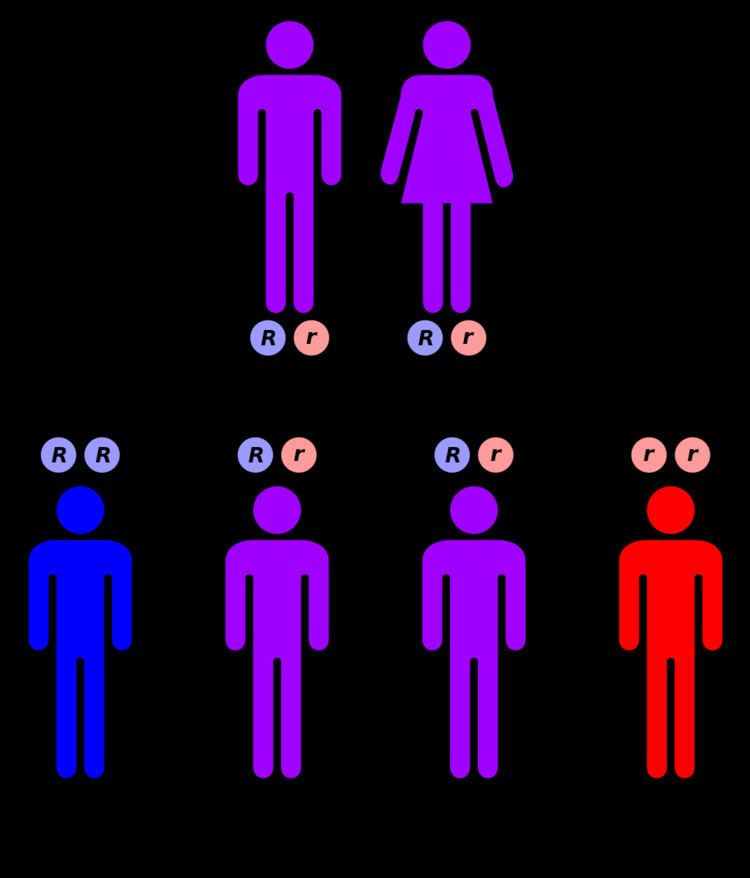
Sanfilippo syndrome, or mucopolysaccharidosis III (MPS-III) is a rare autosomal recessive lysosomal storage disease. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan heparan sulfate (which is found in the extra-cellular matrix and on cell surface glycoproteins).
Contents
Although undegraded heparan sulfate is the primary stored substrate, glycolipids such as gangliosides are also stored despite no genetic defect in the enzymes associated with their breakdown.
The condition is named for Sylvester Sanfilippo, the pediatrician who first described the disease.
Incidence
Incidence of Sanfilippo syndrome varies geographically, with approximately 1 case per 280,000 live births in Northern Ireland, 1 per 66,000 in Australia, and 1 per 50,000 in the Netherlands.
The Australian study estimated the following incidences for each subtype of Sanfilippo syndrome:
Pathophysiology
The four types of MPS-III are due to specific enzyme deficiencies affecting the breakdown of heparan sulfate, which then builds up in various organs. All four types have autosomal recessive inheritance.
Diagnosis and natural history
MPS-III A, B, C and D are considered to be clinically indistinguishable, although mutations in different genes are responsible for each disease. The following discussion is therefore applicable to all four conditions.
The disease manifests in young children. Affected infants are apparently normal, although some mild facial dysmorphism may be noticeable. The stiff joints, hirsuteness and coarse hair typical of other mucopolysaccharidoses are usually not present until late in the disease. After an initial symptom-free interval, patients usually present with a slowing of development and/or behavioral problems, followed by progressive intellectual decline resulting in severe dementia and progressive motor disease. Acquisition of speech is often slow and incomplete. The disease progresses to increasing behavioural disturbance including temper tantrums, hyperactivity, destructiveness, aggressive behaviour, pica and sleep disturbance. As affected children have normal muscle strength and mobility, the behavioural disturbances are very difficult to manage. The disordered sleep in particular presents a significant problem to care providers. In the final phase of the illness, children become increasingly immobile and unresponsive, often require wheelchairs, and develop swallowing difficulties and seizures. The life-span of an affected child does not usually extend beyond late teens to early twenties.
Although the clinical features of the disease are mainly neurological, patients may also develop diarrhea, carious teeth, and an enlarged liver and spleen. There is a broad range of clinical severity. The disease may very rarely present later in life as a psychotic episode.
Of all the MPS diseases, MPS III produces the mildest physical abnormalities. It is important, however, that simple and treatable conditions such as ear infections and toothaches not be overlooked because of behavior problems that make examination difficult. Children with MPS III often have an increased tolerance of pain. Bumps and bruises or ear infections that would be painful for other children often go unnoticed in children with MPS III. Parents may need to search for a doctor with the patience and interest in treating a child with a long-term illness. Some children with MPS III may have a blood-clotting problem during and after surgery. [1]
The diagnosis may be confirmed by assay of enzyme levels in tissue samples and gene sequencing. Prenatal diagnosis is possible.
Treatment
Treatment remains largely supportive. The behavioral disturbances of MPS-III respond poorly to medication. If an early diagnosis is made, bone marrow replacement may be beneficial. Although the missing enzyme can be manufactured and given intravenously, it cannot penetrate the blood–brain barrier and therefore cannot treat the neurological manifestations of the disease.
Along with many other lysosomal storage diseases, MPS-III exists as a model of a monogenetic disease involving the central nervous system. Several promising therapies are in development. Gene therapy in particular is under Phase I/II clinical trial in France since October 2011 under the leadership of Paris-based biotechnology company Lysogene [2]. Other potential therapies include chemical modification of deficient enzymes to allow them to penetrate the blood–brain barrier, stabilisation of abnormal but active enzyme to prevent its degradation, and implantation of stem cells strongly expressing the missing enzyme. For any future treatment to be successful, it must be administered as early as possible. Currently MPS-III is mainly diagnosed clinically, by which stage it is probably too late for any treatment to be very effective. Neonatal screening programs would provide the earliest possible diagnosis.
The flavonoid genistein decreases the pathological accumulation of glycosaminoglycans in Sanfilippo syndrome. In vitro, animal studies and clinical experiments suggest that the symptoms of the disease may be alleviated by an adequate dose of genistein. Despite its reported beneficial properties, genistein also has toxic side effects.
Several US-based groups have recently been organized to speed the development of new treatments for Sanfilippo syndrome.