
 | ||
Rubber elasticity, a well-known example of hyperelasticity, describes the mechanical behavior of many polymers, especially those with cross-links.
Contents
- Thermodynamics
- Models
- Freely jointed chain model
- Worm like chain model
- Integrated Rubber Network Models
- The Molecular Kink Paradigm for Rubber Elasticity
- Low chain extension regime Ia
- Moderate chain extension regime Ib
- High chain extension regime II
- Network morphology
- Numerical network simulation model
- History
- References
Thermodynamics
Temperature affects the elasticity of elastomers in an unusual way. When the elastomer is assumed to be in a stretched state, heating causes them to contract. Vice versa, cooling can cause expansion. This can be observed with an ordinary rubber band. Stretching a rubber band will cause it to release heat (press it against your lips), while releasing it after it has been stretched will lead it to absorb heat, causing its surroundings to become cooler. This phenomenon can be explained with the Gibbs Free Energy. Rearranging ΔG=ΔH−TΔS, where G is the free energy, H is the enthalpy, and S is the entropy, we get TΔS=ΔH−ΔG. Since stretching is nonspontaneous, as it requires external work, TΔS must be negative. Since T is always positive (it can never reach absolute zero), the ΔS must be negative, implying that the rubber in its natural state is more entangled (with more microstates) than when it is under tension. Thus, when the tension is removed, the reaction is spontaneous, leading ΔG to be negative. Consequently, the cooling effect must result in a positive ΔH, so ΔS will be positive there.
The result is that an elastomer behaves somewhat like an ideal monatomic gas, inasmuch as (to good approximation) elastic polymers do not store any potential energy in stretched chemical bonds or elastic work done in stretching molecules, when work is done upon them. Instead, all work done on the rubber is "released" (not stored) and appears immediately in the polymer as thermal energy. In the same way, all work that the elastic does on the surroundings results in the disappearance of thermal energy in order to do the work (the elastic band grows cooler, like an expanding gas). This last phenomenon is the critical clue that the ability of an elastomer to do work depends (as with an ideal gas) only on entropy-change considerations, and not on any stored (i.e., potential) energy within the polymer bonds. Instead, the energy to do work comes entirely from thermal energy, and (as in the case of an expanding ideal gas) only the positive entropy change of the polymer allows its internal thermal energy to be converted efficiently (100% in theory) into work.
Models
Invoking the theory of rubber elasticity, one considers a polymer chain in a crosslinked network as an entropic spring. When the chain is stretched, the entropy is reduced by a large margin because there are fewer conformations available. Therefore, there is a restoring force, which causes the polymer chain to return to its equilibrium or unstretched state, such as a high entropy random coil configuration, once the external force is removed. This is the reason why rubber bands return to their original state. Two common models for rubber elasticity are the freely-jointed chain model and the worm-like chain model.
Freely-jointed chain model
Polymers can be modeled as freely jointed chains with one fixed end and one free end (FJC model):
where
Note that the movement could be backwards or forwards, so the net time average
Ideally, the polymer chain's movement is purely entropic (no enthalpic, or heat-related, forces involved). By using the following basic equations for entropy and Helmholtz free energy, we can model the driving force of entropy "pulling" the polymer into an unstretched conformation. Note that the force equation resembles that of a spring: F=kx.
Note that the elastic coefficient
Worm-like chain model
The worm-like chain model (WLC) takes the energy required to bend a molecule into account. The variables are the same except that
Therefore, when there is no distance between chain ends (r=0), the force required to do so is zero, and to fully extend the polymer chain (
Integrated Rubber Network Models
Following its introduction to Europe from the New World in the late 15th century, rubber was regarded mostly as a fascinating curiosity until 1838, when the American inventor Charles Goodyear found that its properties could be immensley improved by adding a few percent sulphur and heating. What he produced was a rubber network; the short sulphur chains produced covalent crosslinks between adjacent polymer chains in the liquid (melt) rubber, essentially transforming the sample into a single molecule. The network is the sine qua non of polymer elastomers. To study the mechanical properties of rubber requires not only chain force-extension models but also a method to account for the geometric effects of the network, specifically, how the chain end-to-end distance changes with a macroscopic tensile strain (the ratio of the increase in length to the original length). Historically, elasticity theories began with the ansatz that a volume element of a rubber network could be represented by a single cross-link node as a connection point for a few chains. Early versions combined a chain force model (such as the Freely jointed chain model) with a simple network model that consisted of a cross-link node with 3 or more equal chains having orthogonal end-to-end vectors, oriented symmetrically with the strain axis. To relate the network chain extension to the macroscopic strain, it was assumed that the cross-link node coordinates undergo an affine transformation with respect to the applied strain. With these assumptions, formulas could be derived for the macroscopic stress vs. strain. A new theory of rubber elasticity, 'The Molecular Kink Paradigm' has recently been introduced that associates elastic chain forces with molecule-specific physical mechanisms (entropic and enthalpic) that occur as a network chain is put in tension. The theory also includes an explicit polymer network model that captures the complex morphology of a rubber network, including chain rupture and network failure.
The Molecular Kink Paradigm for Rubber Elasticity
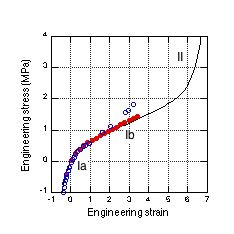
The Molecular Kink Paradigm proceeds from the intuitive notion that the chains that make up a natural rubber (polyisoprene) network are constrained by surrounding chains to remain within a ‘tube’, and that elastic forces produced in a chain, as a result of some applied strain, are propagated along the chain contour within this tube. Over experimental time scales, only short sections of the chain, comprised of a few backbone units, are free to occupy all allowed rotational conformations as given by an equilibrium Boltzmann distribution. Changes in the entropy of a chain are then associated with the thermal motion of short regions that can move more or less freely within the tube. These non-straight regions evoke the concept of ‘kinks’ that in fact manifest the random-walk nature of the chain. As a network is subjected to strain, some kinks are forced into more extended conformations, causing a decrease in entropy that produces an elastic force along the chain. There are three distinct molecular mechanisms that produce these forces, two of which arise from changes in entropy that we shall refer to as low chain extension regime, Ia and moderate chain extension regime, Ib. The third mechanism occurs at high chain extension, as it is extended beyond its initial equilibrium contour length by the distortion of the chemical bonds along its backbone. In this case, the restoring force is spring-like and we shall refer to it as regime II. The three force mechanisms are found to roughly correspond to the three regions observed in tensile stress vs. strain experiments, shown in Fig. 1.
All of these chain force models are non-zero in extension only, i.e., the force required to extend a chain is assumed to be zero unless the chain end-to-end distance is increased.
The initial morphology of the network, immediately after chemical cross-linking, is governed by two random processes: (1) The probability for a cross-link to occur at any isoprene unit and, (2) the random walk nature of the chain conformation. The end-to-end distance probability distribution for a fixed chain length, i.e. fixed number of isoprene units, is described by a random walk. It is the joint probability distribution of the network chain lengths and the end-to-end distances between their cross-link nodes that characterizes the network morphology. Because both the molecular physics mechanisms that produce the elastic forces and the complex morphology of the network must be treated simultaneously, simple analytic elasticity models are not possible; an explicit 3-dimensional numerical model is required to simulate the effects of strain on a representative volume element of a network.
Low chain extension regime, Ia
At very low strain, the molecular mechanism for elasticity arises from the distortion, or stretching, of kinks along the chain contour. Physically, the applied strain causes the kinks to be forced beyond from their thermal equilibrium end-to-end distances. A force constant for this regime can be estimated by sampling molecular dynamics (MD) trajectories of short chains. From these MD trajectories, the probability distributions of end-to-end distance for short kinks, comprised of 2-4 isoprene units, can be obtained. Since these distributions (which turns out to be approximately Gaussian) are directly related to the number of states at each distance, we may associate them with an entropy change of the kink. By numerically differentiating the probability distribution, the change in entropy, and hence free energy, with respect to the kink end-to-end distance can be found. The force model for this regime is linear and proportional to the temperature divided by the chain tortuosity (the ratio of the chain contour length divided by its end-to-end distance).
Moderate chain extension regime, Ib
The physical process that gives rise to the elastic force in the moderate chain extension regime is the gradual straightening of the chain. At full chain extension, (i.e., the onset of regime II), the applied tension forces all of the isoprene units along the chain backbone to lie along piece-wise straight lines. Numerous experiments strongly suggest that the molecular mechanism responsible for the elastic force must be associated with a change in chain entropy. How does a chain become straight? From MD simulations of free natural rubber molecules at temperatures near 300 K, we can study the conformations of the chain backbone. We find that departures of the chain backbone from linearity occur over contour lengths of just a few isoprene units (a kink).
Although an isoprene unit (Fig. 2) is free to rotate about each of its single C-C bonds, there are typically 3 favored rotational conformations, separated by ~120 degrees, that correspond to energy minima. An isoprene unit has three single C-C bonds and 18 allowed rotational conformations, each one with a unique end-to-end distance and energy. States with shorter end-to-end distances tend to have a higher energy. We designate these as ‘compact’ states and those having greater end-to-end distances as ‘extended’. As a network chain is gradually extended toward linear (but still confined by a surrounding tube), kinks must be straightened. Six of the 18 isoprene rotational conformations are extended states and, as the chain is straightened, more and more of the isoprene units are forced to spend more time these states. It is the decrease in entropy associated with reducing the number of rotational states allowed for each isoprene unit that gives rise to the elastic force in this regime. A force constant for chain extension can be estimated from the change in free energy associated with the entropy change that occurs as the occupancy of some rotational states is decreased. As with regime Ia, the force model for this regime is linear and proportional to the temperature divided by the chain tortuosity (the ratio of the chain contour length divided by its end-to-end distance). As the chain is extended, the tortuosity decreases from its initial value to 1.
High chain extension regime, II
When a rubber sample is stretched sufficiently far, we know from experience that it breaks more or less cleanly in a plane perpendicular to the strain axis. It follows that covalent bonds on network chains must undergo a bond rupture as a consequence of the imposed strain. Some network chains can rupture before the entire sample completely fails but, as more and more chains break, too few network chains remain intact to support the imposed tensile stress, causing the sample to abruptly fail. The intrinsic molecular mechanisms that give rise to the strong elastic chain force in this region are bond distortions, e.g., bond angle increases, bond stretches and dihedral angle rotations. These forces are spring-like and are not associated with entropy changes. The tensile force along a chain required to cause bond rupture has been calculated via quantum chemistry simulations and it is approximately 7 nN, about a factor of a thousand greater than the entropic chain forces at low strain. The angles between adjacent backbone C-C bonds in an isoprene unit vary between about 115-120 degrees and the forces associated with maintaining these angles are quite large, so within each unit, the chain backbone always follows a zigzag path. The same quantum chemistry simulations also predict that a natural rubber chain can be stretched by about 40% beyond its sensibly-straight state before rupture, and also provide a force extension curve (fit to a fifth order polynomial) that can be used in a numerical network model. The steep upturn in the elastic stress, observed at moderate to high strains (Fig. 2), is due to the extension of network chains beyond their sensibly-straight state.
Network morphology
The initial morphology of the network is dictated by two random processes: the probability for a crosslink to occur at any isoprene unit and the Markov random walk nature of a chain conformation. The end-to-end distance distribution for a fixed chain length is generated by a Markov sequence. This conditional probability density function relates the chain length
The probability that any isoprene unit becomes part of a cross-link node is proportional to the ratio of the concentrations of the cross-linker molecules (e.g., dicumyl-peroxide) to the isoprene units:
The factor of two comes about because two isoprene units (one from each chain) participate in the crosslink. The probability for finding a chain containing
where
The complex morphology of a natural rubber network can be seen in Fig. 3, which shows the probability density vs. end-to-end distance (in units of mean node spacing) for an ‘average’ chain. For the common experimental cross-link density of 4x1019 cm−3, an average chain contains about 116 isoprene units (52 Kuhn lengths) and has a contour length of about 50 nm. Fig. 3 shows that a significant fraction of chains span several node spacings, i.e., the chain ends overlap other network chains. As the network is strained, paths composed of these more extended chains emerge that span the entire sample, and it is these paths that carry most of the stress at high strains.
Numerical network simulation model
To calculate the elastic response of a rubber sample, the three chain force models (regimes Ia, Ib and II) and the network morphology must be combined in a micro-mechanical network model. Using the joint probability distribution in equation (4) and the force extension models, it is possible to devise numerical algorithms to both construct a faithful representative volume element of a network and to simulate the resulting mechanical stress as it is subjected to strain. An iterative relaxation algorithm is used to maintain approximate force equilibrium at each network node as strain is imposed. When the force constant obtained for kinks having 2 or 3 isoprene units (approximately one Kuhn length) is used in numerical simulations, the predicted stress is found to be consistent with experiments. The results of such a calculation are shown in Fig. 1 as a solid blue line. These simulations also predict a steep upturn in the stress as network chains are forced into extension regime II and, ultimately, material failure due to bond rupture.
History
Eugene Guth and Hubert M. James proposed the entropic origins of rubber elasticity in 1941.