
 | ||
In medicine, proteopathy (Proteo- [pref. protein]; -pathy [suff. disease]; proteopathies pl.; proteopathic adj.) refers to a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body. Often the proteins fail to fold into their normal configuration; in this misfolded state, the proteins can become toxic in some way (a gain of toxic function) or they can lose their normal function. The proteopathies (also known as proteinopathies, protein conformational disorders, or protein misfolding diseases) include such diseases as Creutzfeldt–Jakob disease, Alzheimer's disease, Parkinson's disease, prion disease, amyloidosis, and a wide range of other disorders (see List of Proteopathies).
Contents
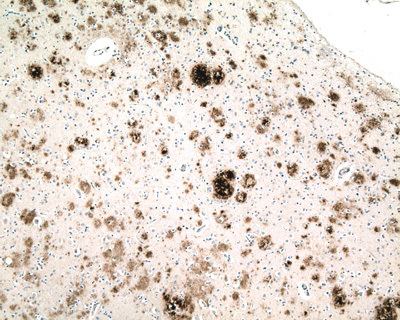
The concept of proteopathy can trace its origins to the mid-19th century, when, in 1854, Rudolf Virchow coined the term amyloid ("starch-like") to describe a substance in cerebral corpora amylacea that exhibited a chemical reaction resembling that of cellulose. In 1859, Friedreich and Kekulé demonstrated that, rather than consisting of cellulose, "amyloid" actually is rich in protein. Subsequent research has shown that many different proteins can form amyloid, and that all amyloids have in common birefringence in cross-polarized light after staining with the dye Congo Red, as well as a fibrillar ultrastructure when viewed with an electron microscope. However, some proteinaceous lesions lack birefringence and contain few or no classical amyloid fibrils, such as the diffuse deposits of Aβ protein in the brains of Alzheimer patients. Furthermore, evidence has emerged that small, non-fibrillar protein aggregates known as oligomers are toxic to the cells of an affected organ, and that amyloidogenic proteins in their fibrillar form may be relatively benign.
Pathophysiology
In most, if not all proteopathies, a change in 3-dimensional folding (conformation) increases the tendency of a specific protein to bind to itself. In this aggregated form, the protein is resistant to clearance and can interfere with the normal capacity of the affected organs. In some cases, misfolding of the protein results in a loss of its usual function. For example, cystic fibrosis is caused by a defective cystic fibrosis transmembrane conductance regulator (CFTR) protein, and in amyotrophic lateral sclerosis / frontotemporal lobar degeneration (FTLD), certain gene-regulating proteins inappropriately aggregate in the cytoplasm, and thus are unable to perform their normal tasks within the nucleus. Because proteins share a common structural feature known as the polypeptide backbone, all proteins have the potential to misfold under some circumstances. However, only a relatively small number of proteins are linked to proteopathic disorders, possibly due to structural idiosyncrasies of the vulnerable proteins. For example, proteins that are relatively unstable as monomers (that is, as single, unbound protein molecules) are more likely to misfold into an abnormal conformation. In nearly all instances, the disease-causing molecular configuration involves an increase in beta-sheet secondary structure of the protein. The abnormal proteins in some proteopathies have been shown to fold into multiple 3-dimensional shapes; these variant, proteinaceous structures are defined by their different pathogenic, biochemical, and conformational properties. They have been most thoroughly studied with regard to prion disease, and are referred to as protein strains.
The likelihood that proteopathy will develop is increased by certain risk factors that promote the self-assembly of a protein. These include destabilizing changes in the primary amino acid sequence of the protein, post-translational modifications (such as hyperphosphorylation), changes in temperature or pH, an increase in production of a protein, or a decrease in its clearance. Advancing age is a strong risk factor, as is traumatic brain injury. In the aging brain, multiple proteopathies can overlap. For example, in addition to tauopathy and Aβ-amyloidosis (which coexist as key pathologic features of Alzheimer's disease), many Alzheimer patients have concomitant synucleinopathy (Lewy bodies) in the brain.
It is hypothesized that chaperones and co-chaperones (proteins that assist protein folding) may antagonize proteotoxicity during aging and in protein misfolding-diseases to maintain proteostasis.
Seeded induction
Some proteins can be induced to form abnormal assemblies by exposure to the same (or similar) protein that has folded into a disease-causing conformation, a process called 'seeding' or 'permissive templating'. In this way, the disease state can be brought about in a susceptible host by the introduction of diseased tissue extract from an afflicted donor. The best known form of such inducible proteopathy is prion disease, which can be transmitted by exposure of a host organism to purified prion protein in a disease-causing conformation. There is now evidence that other proteopathies can be induced by a similar mechanism, including Aβ amyloidosis, amyloid A (AA) amyloidosis, and apolipoprotein AII amyloidosis, tauopathy, synucleinopathy, and the aggregation of superoxide dismutase-1 (SOD1), polyglutamine, and TAR DNA-binding protein-43 (TDP-43).
In all of these instances, an aberrant form of the protein itself appears to be the pathogenic agent. In some cases, the deposition of one type of protein can be experimentally induced by aggregated assemblies of other proteins that are rich in β-sheet structure, possibly because of structural complementarity of the protein molecules. For example, AA amyloidosis can be stimulated in mice by such diverse macromolecules as silk, the yeast amyloid Sup35, and curli from the bacterium Escherichia coli. In addition, apolipoprotein AII amyloid can be induced in mice by a variety of β-sheet rich amyloid fibrils, and cerebral tauopathy can be induced by brain extracts that are rich in aggregated Aβ. There is also experimental evidence for cross-seeding between prion protein and Aβ. In general, such heterologous seeding is less efficient than is seeding by a corrupted form of the same protein.