
Specialty infectious disease ICD-9-CM 046 | ICD-10 A81 | |
 | ||
A prion is an infectious agent composed entirely of protein material, called PrP (short for prion protein), that can fold in multiple, structurally distinct ways, at least one of which is transmissible to other prion proteins, leading to disease that is similar to viral infection. They are suspected to be the cause of transmissible spongiform encephalopathies (TSEs) among other diseases.
Contents
- Discovery
- Structure
- PrPC
- PrPres
- PrPSc
- Function
- PrP and long term memory
- PrP and stem cell renewal
- Prion replication mechanism
- Prion diseases and their transmission properties
- Transmission
- Prions in plants
- Sterilization
- Prion like domains
- Role in neurodegenerative disease
- Fungi
- Potential treatments and diagnosis
- Causal hypotheses
- Protein only hypothesis
- Genetic factors
- Multi component hypothesis
- Heavy metal poisoning hypothesis
- Viral hypothesis
- Virino hypothesis
- Spiroplasma hypothesis
- Acinetobacter autoimmunity hypothesis
- Etymology and pronunciation
- References
Prions were initially identified as the causative agent in animal TSEs such as bovine spongiform encephalopathy (BSE)—known popularly as "mad cow disease"—and scrapie in sheep. Human prion diseases include Creutzfeldt–Jakob disease (CJD) and its variant (vCJD), Gerstmann–Sträussler–Scheinker syndrome, fatal familial insomnia, and kuru. A 2015 study concluded that multiple system atrophy (MSA), a rare human neurodegenerative disease, is caused by a misfolded version of a protein called alpha-synuclein, and is therefore also classifiable as a prion disease. Several yeast proteins have been identified as having prionogenic properties as well.
A protein as a standalone infectious agent stands in contrast to all other known infectious agents such as viruses, bacteria, fungi, and parasites, all of which contain nucleic acids (DNA, RNA, or both). For this reason, a minority of researchers still consider the prion/TSE hypothesis unproven. All known prion diseases in mammals affect the structure of the brain or other neural tissue. No effective medical treatment is known. The illness is progressive and always fatal.
Prions may propagate by transmitting their misfolded protein state. When a prion enters a healthy organism, it induces existing, properly folded proteins to convert into the misfolded prion form. In this way, the prion acts as a template to guide the misfolding of more proteins into prion form. In yeast, this refolding is assisted by chaperone proteins such as Hsp104p. These refolded prions can then go on to convert more proteins themselves, leading to a chain reaction resulting in large amounts of the prion form. All known prions induce the formation of an amyloid fold, in which the protein polymerises into an aggregate consisting of tightly packed beta sheets. Amyloid aggregates are fibrils, growing at their ends, and replicate when breakage causes two growing ends to become four growing ends. The incubation period of prion diseases is determined by the exponential growth rate associated with prion replication, which is a balance between the linear growth and the breakage of aggregates. The propagation of the prion depends on the presence of normally folded protein in which the prion can induce misfolding; animals that do not express the normal form of the prion protein can neither develop nor transmit the disease.
Prion aggregates are extremely stable and accumulate in infected tissue, causing tissue damage and cell death. This structural stability means that prions are resistant to denaturation by chemical and physical agents, making disposal and containment of these particles difficult. Prion structure varies slightly between species, but nonetheless prion replication is subject to occasional epimutation and natural selection just like other forms of replication.
Discovery
During the 1960s, two London-based researchers, radiation biologist Tikvah Alper and biophysicist John Stanley Griffith, developed the hypothesis that some transmissible spongiform encephalopathies are caused by an infectious agent consisting solely of proteins. Earlier investigations by E. J. Field into scrapie and kuru had identified the transfer of pathologically inert polysaccharides that only become infectious in the host. Alper and Griffith wanted to account for the discovery that the mysterious infectious agent causing the diseases scrapie and Creutzfeldt–Jakob disease resisted ionizing radiation. (A single ionizing "hit" normally destroys an entire infectious particle, and the dose needed to hit half the particles depends on the size of the particles. Empirical results of ionizing doses applied to the unknown infectious substance evidenced an infectious particle size too small to be a viral mechanism.)
Francis Crick recognized the potential importance of the Griffith protein-only hypothesis for scrapie propagation in the second edition of his "Central dogma of molecular biology" (1970): While asserting that the flow of sequence information from protein to protein, or from protein to RNA and DNA was "precluded", he noted that Griffith's hypothesis was a potential contradiction (although it was not so promoted by Griffith). The revised hypothesis was later formulated, in part, to accommodate reverse transcription (which both Howard Temin and David Baltimore discovered in 1970).
In 1982, Stanley B. Prusiner of the University of California, San Francisco announced that his team had purified the hypothetical infectious prion, and that the infectious agent consisted mainly of a specific protein – though they did not manage to isolate the protein until two years after Prusiner's announcement. While the infectious agent was named a prion, the specific protein that the prion was composed of is also known as the Prion Protein (PrP), though this protein may occur both in infectious and non-infectious forms. Prusiner won the Nobel Prize in Physiology or Medicine in 1997 for his research into prions.
Structure
The protein that prions are made of (PrP) is found throughout the body, even in healthy people and animals. However, PrP found in infectious material has a different structure and is resistant to proteases, the enzymes in the body that can normally break down proteins. The normal form of the protein is called PrPC, while the infectious form is called PrPSc — the C refers to 'cellular' PrP, while the Sc refers to 'scrapie', the prototypic prion disease, occurring in sheep. While PrPC is structurally well-defined, PrPSc is certainly polydisperse and defined at a relatively poor level. PrP can be induced to fold into other more-or-less well-defined isoforms in vitro, and their relationship to the form(s) that are pathogenic in vivo is not yet clear.
PrPC
PrPC is a normal protein found on the membranes of cells. It has 209 amino acids (in humans), one disulfide bond, a molecular mass of 35–36 kDa and a mainly alpha-helical structure. Several topological forms exist; one cell surface form anchored via glycolipid and two transmembrane forms. The normal protein is not sedimentable; meaning that it cannot be separated by centrifuging techniques. Its function is a complex issue that continues to be investigated. PrPC binds copper (II) ions with high affinity. The significance of this finding is not clear, but it is presumed to relate to PrP structure or function. PrPC is readily digested by proteinase K and can be liberated from the cell surface in vitro by the enzyme phosphoinositide phospholipase C (PI-PLC), which cleaves the glycophosphatidylinositol (GPI) glycolipid anchor. PrP has been reported to play important roles in cell-cell adhesion and intracellular signaling in vivo, and may therefore be involved in cell-cell communication in the brain.
PrPres
Protease-resistant PrPSc-like protein (PrPres) is an isoform of PrPc from which is structurally altered and converted into a misfolded proteinase K-resistant form in vitro. To model conversion of PrPC to PrPSc in vitro, Saborio et al. rapidly converted PrPC into a PrPres by a procedure involving cyclic amplification of protein misfolding. The term "PrPres" has been made to distinguish between PrPSc, which is isolated from infectious tissue and associated with the transmissible spongiform encephalopathy agent. For example, unlike PrPsc, PrPres may not necessarily be infectious.
PrPSc
The infectious isoform of PrP, known as PrPSc, is able to convert normal PrPC proteins into the infectious isoform by changing their conformation, or shape; this, in turn, alters the way the proteins interconnect. PrPSc always causes prion disease. Although the exact 3D structure of PrPSc is not known, it has a higher proportion of β-sheet structure in place of the normal α-helix structure. Aggregations of these abnormal isoforms form highly structured amyloid fibers, which accumulate to form plaques. It is unclear as to whether these aggregates are the cause of cell damage or are simply a side-effect of the underlying disease process. The end of each fiber acts as a template onto which free protein molecules may attach, allowing the fiber to grow. Under most circumstances, only PrP molecules with an identical amino acid sequence to the infectious PrPSc are incorporated into the growing fiber. However, rare cross-species transmission is also possible.
Function
The physiological function of the prion protein remains a controversial matter. While data from in vitro experiments suggest many dissimilar roles, studies on PrP knockout mice have provided only limited information because these animals exhibit only minor abnormalities. In research done in mice, it was found that the cleavage of PrP proteins in peripheral nerves causes the activation of myelin repair in Schwann Cells and that the lack of PrP proteins caused demyelination in those cells.
PrP and long-term memory
A review of evidence in 2005 suggested that PrP may have a normal function in maintenance of long-term memory. As well, a 2004 study found that mice lacking genes for normal cellular PrP protein show altered hippocampal long-term potentiation.
PrP and stem cell renewal
A 2006 article from the Whitehead Institute for Biomedical Research indicates that PrP expression on stem cells is necessary for an organism's self-renewal of bone marrow. The study showed that all long-term hematopoietic stem cells express PrP on their cell membrane and that hematopoietic tissues with PrP-null stem cells exhibit increased sensitivity to cell depletion.
Prion replication mechanism
The first hypothesis that tried to explain how prions replicate in a protein-only manner was the heterodimer model. This model assumed that a single PrPSc molecule binds to a single PrPC molecule and catalyzes its conversion into PrPSc. The two PrPSc molecules then come apart and can go on to convert more PrPC. However, a model of prion replication must explain both how prions propagate, and why their spontaneous appearance is so rare. Manfred Eigen showed that the heterodimer model requires PrPSc to be an extraordinarily effective catalyst, increasing the rate of the conversion reaction by a factor of around 1015. This problem does not arise if PrPSc exists only in aggregated forms such as amyloid, where cooperativity may act as a barrier to spontaneous conversion. What is more, despite considerable effort, infectious monomeric PrPSc has never been isolated.
An alternative model assumes that PrPSc exists only as fibrils, and that fibril ends bind PrPC and convert it into PrPSc. If this were all, then the quantity of prions would increase linearly, forming ever longer fibrils. But exponential growth of both PrPSc and of the quantity of infectious particles is observed during prion disease. This can be explained by taking into account fibril breakage. A mathematical solution for the exponential growth rate resulting from the combination of fibril growth and fibril breakage has been found. The exponential growth rate depends largely on the square root of the PrPC concentration. The incubation period is determined by the exponential growth rate, and in vivo data on prion diseases in transgenic mice match this prediction. The same square root dependence is also seen in vitro in experiments with a variety of different amyloid proteins.
The mechanism of prion replication has implications for designing drugs. Since the incubation period of prion diseases is so long, an effective drug does not need to eliminate all prions, but simply needs to slow down the rate of exponential growth. Models predict that the most effective way to achieve this, using a drug with the lowest possible dose, is to find a drug that binds to fibril ends and blocks them from growing any further.
Prion diseases and their transmission properties
Until 2015, all known mammalian prion diseases were caused by the so-called prion protein, PrP, when Multiple System Atrophy was found to be likely caused by a new prion called Alpha-synuclein, The endogenous, properly folded form is denoted PrPC (for Common or Cellular), whereas the disease-linked, misfolded form is denoted PrPSc (for Scrapie, after one of the diseases first linked to prions and neurodegeneration.) The precise structure of the prion is not known, though they can be formed by combining PrPC, polyadenylic acid, and lipids in a Protein Misfolding Cyclic Amplification (PMCA) reaction. Proteins showing prion-type behavior are also found in some fungi, which has been useful in helping to understand mammalian prions. Fungal prions do not appear to cause disease in their hosts.
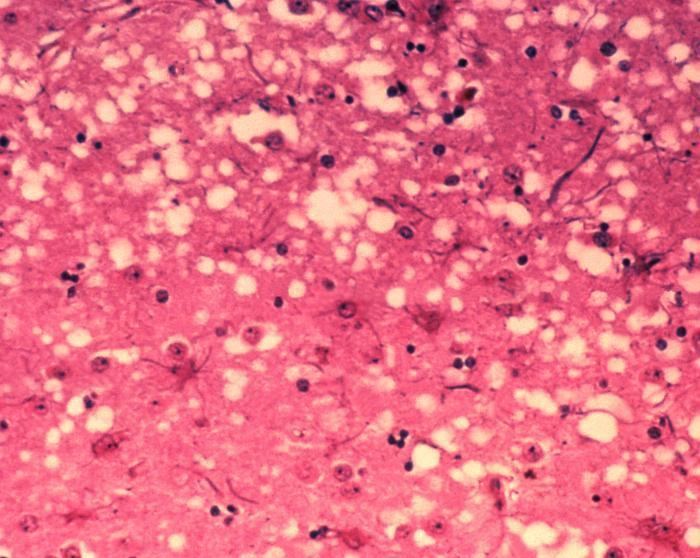
Prions cause neurodegenerative disease by aggregating extracellularly within the central nervous system to form plaques known as amyloid, which disrupt the normal tissue structure. This disruption is characterized by "holes" in the tissue with resultant spongy architecture due to the vacuole formation in the neurons. Other histological changes include astrogliosis and the absence of an inflammatory reaction. While the incubation period for prion diseases is relatively long (5 to 20 years), once symptoms appear the disease progresses rapidly, leading to brain damage and death. Neurodegenerative symptoms can include convulsions, dementia, ataxia (balance and coordination dysfunction), and behavioural or personality changes.
All known prion diseases, collectively called transmissible spongiform encephalopathies (TSEs), are untreatable and fatal. However, a vaccine developed in mice may provide insight into providing a vaccine to resist prion infections in humans. Additionally, in 2006 scientists announced that they had genetically engineered cattle lacking a necessary gene for prion production – thus theoretically making them immune to BSE, building on research indicating that mice lacking normally occurring prion protein are resistant to infection by scrapie prion protein. In 2013, a study revealed that 1 in 2,000 people in the United Kingdom might harbour the infectious prion protein that causes vCJD.
Many different mammalian species can be affected by prion diseases, as the prion protein (PrP) is very similar in all mammals. Due to small differences in PrP between different species it is unusual for a prion disease to transmit from one species to another. The human prion disease variant Creutzfeldt–Jakob disease, however, is believed to be caused by a prion that typically infects cattle, causing Bovine spongiform encephalopathy and is transmitted through infected meat.
Transmission
It has been recognized that prion diseases can arise in three different ways: acquired, familial, or sporadic. It is often assumed that the diseased form directly interacts with the normal form to make it rearrange its structure. One idea, the "Protein X" hypothesis, is that an as-yet unidentified cellular protein (Protein X) enables the conversion of PrPC to PrPSc by bringing a molecule of each of the two together into a complex.
Current research suggests that the primary method of infection in animals is through ingestion. It is thought that prions may be deposited in the environment through the remains of dead animals and via urine, saliva, and other body fluids. They may then linger in the soil by binding to clay and other minerals.
A University of California research team, led by Nobel Prize winner Stanley Prusiner, has provided evidence for the theory that infection can occur from prions in manure. And, since manure is present in many areas surrounding water reservoirs, as well as used on many crop fields, it raises the possibility of widespread transmission. It was reported in January 2011 that researchers had discovered prions spreading through airborne transmission on aerosol particles, in an animal testing experiment focusing on scrapie infection in laboratory mice. Preliminary evidence supporting the notion that prions can be transmitted through use of urine-derived human menopausal gonadotropin, administered for the treatment of infertility, was published in 2011.
Prions in plants
In 2015, researchers at The University of Texas Health Science Center at Houston found that plants can be a vector for prions. When researchers fed hamsters grass that grew on ground where a deer that died with chronic wasting disease (CWD) was buried, the hamsters became ill with CWD, suggesting that prions can bind to plants, which then take them up into the leaf and stem structure, where they can be eaten by herbivores, thus completing the cycle. It is thus possible that there is a progressively accumulating number of prions in the environment.
Sterilization
Infectious particles possessing nucleic acid are dependent upon it to direct their continued replication. Prions, however, are infectious by their effect on normal versions of the protein. Sterilizing prions, therefore, requires the denaturation of the protein to a state in which the molecule is no longer able to induce the abnormal folding of normal proteins. In general, prions are quite resistant to proteases, heat, ionizing radiation, and formaldehyde treatments, although their infectivity can be reduced by such treatments. Effective prion decontamination relies upon protein hydrolysis or reduction or destruction of protein tertiary structure. Examples include sodium hypochlorite, sodium hydroxide, and strongly acidic detergents such as LpH. 134 °C (274 °F) for 18 minutes in a pressurized steam autoclave has been found to be somewhat effective in deactivating the agent of disease. Ozone sterilization is currently being studied as a potential method for prion denaturation and deactivation. Renaturation of a completely denatured prion to infectious status has not yet been achieved; however, partially denatured prions can be renatured to an infective status under certain artificial conditions.
The World Health Organization recommends any of the following three procedures for the sterilization of all heat-resistant surgical instruments to ensure that they are not contaminated with prions:
- Immerse in 1N sodium hydroxide and place in a gravity-displacement autoclave at 121 °C for 30 minutes; clean; rinse in water; and then perform routine sterilization processes.
- Immerse in 1N sodium hypochlorite (20,000 parts per million available chlorine) for 1 hour; transfer instruments to water; heat in a gravity-displacement autoclave at 121 °C for 1 hour; clean; and then perform routine sterilization processes.
- Immerse in 1N sodium hydroxide or sodium hypochlorite (20,000 parts per million available chlorine) for 1 hour; remove and rinse in water, then transfer to an open pan and heat in a gravity-displacement (121 °C) or in a porous-load (134 °C) autoclave for 1 hour; clean; and then perform routine sterilization processes.
Prion-like domains
While PrP is considered the only mammalian prion, prion-like domains have been found in a variety of other mammalian proteins. Some of these proteins have been implicated in the ontogeny of age-related neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS, known as Motor Neurone Disease outside the US), frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U), Alzheimer's disease, and Huntington's disease, as well as some forms of Systemic Amyloidosis including AA (Secondary) Amyloidosis that develops in humans and animals with inflammatory and infectious diseases such as Tuberculosis, Crohn's disease, Rheumatoid arthritis, and HIV AIDS. AA amyloidosis, like prion disease, may be transmissible. This has given rise to the 'prion paradigm', where otherwise harmless proteins can be converted to a pathogenic form by a small number of misfolded, nucleating proteins.
The definition of a prion-like domain arises from the study of fungal prions. In yeast, prionogenic proteins have a portable prion domain that is both necessary and sufficient for self-templating and protein aggregation. This has been shown by attaching the prion domain to a reporter protein, which then aggregates like a known prion. Similarly, removing the prion domain from a fungal prion protein inhibits prionogenesis. This modular view of prion behaviour has led to the hypothesis that similar prion domains are present in animal proteins, in addition to PrP. These fungal prion domains have several characteristic sequence features. They are typically enriched in asparagine, glutamine, tyrosine and glycine residues, with an asparagine bias being particularly conducive to the aggregative property of prions. Historically, prionogenesis has been seen as independent of sequence and only dependent on relative residue content. However, this has been shown to be false, with the spacing of prolines and charged residues having been shown to be critical in amyloid formation.
Bioinformatic screens have predicted that over 250 human proteins contain prion-like domains (PrLD). These domains are hypothesized to have the same transmissible, amyloidogenic properties of PrP and known fungal proteins. As in yeast, proteins involved in gene expression and RNA binding seem to be particularly enriched in PrLD's, compared to other classes of protein. In particular, 29 of the known 210 proteins with an RNA recognition motif also have a putative prion domain. Meanwhile, several of these RNA-binding proteins have been independently identified as pathogenic in cases of ALS, FTLD-U, Alzheimer's disease, and Huntington's disease.
Role in neurodegenerative disease
The pathogenicity of prions and proteins with prion-like domains arises from their self-templating ability and the resulting exponential growth of amyloid fibrils. The presence of amyloid fibrils in patients with degenerative diseases has been well documented. These amyloid fibrils are seen as the result of pathogenic proteins that self-propagate and form highly stable, non-functional aggregates. While this does not necessarily imply a causal relationship between amyloid and degenerative diseases, the toxicity of certain amyloid forms and the overproduction of amyloid in familial cases of degenerative disorders supports the idea that amyloid formation is generally toxic.
Specifically, aggregation of TDP-43, an RNA-binding protein, has been found in ALS/MND patients, and mutations in the genes coding for these proteins have been identified in familial cases of ALS/MND. These mutations promote the misfolding of the proteins into a prion-like conformation. The misfolded form of TDP-43 forms cytoplasmic inclusions in afflicted neurons, and is found depleted in the nucleus. In addition to ALS/MND and FTLD-U, TDP-43 pathology is a feature of many cases of Alzheimer's disease, Parkinson's disease and Huntington's disease. The misfolding of TDP-43 is largely directed by its prion-like domain. This domain is inherently prone to misfolding, while pathological mutations in TDP-43 have been found to increase this propensity to misfold, explaining the presence of these mutations in familial cases of ALS/MND. As in yeast, the prion-like domain of TDP-43 has been shown to be both necessary and sufficient for protein misfolding and aggregation.
Similarly, pathogenic mutations have been identified in the prion-like domains of heterogeneous nuclear riboproteins hnRNPA2B1 and hnRNPA1 in familial cases of muscle, brain, bone and motor neuron degeneration. The wild-type form of all of these proteins show a tendency to self-assemble into amyloid fibrils, while the pathogenic mutations exacerbate this behaviour and lead to excess accumulation.
Fungi
Fungal proteins exhibiting templated conformational change were discovered in the yeast Saccharomyces cerevisiae by Reed Wickner in the early 1990s. For their mechanistic similarity to mammalian prions, they were termed yeast prions. Subsequent to this, a prion has also been found in the fungus Podospora anserina. These prions behave similarly to PrP, but, in general, are nontoxic to their hosts. Susan Lindquist's group at the Whitehead Institute has argued some of the fungal prions are not associated with any disease state, but may have a useful role; however, researchers at the NIH have also provided arguments suggesting that fungal prions could be considered a diseased state. There is mounting evidence that fungal proteins have evolved specific functions that are beneficial to the microorganism that enhance their ability to adapt to their diverse environments.
As of 2012, there are eight known prion proteins in fungi, seven in Saccharomyces cerevisiae (Sup35, Rnq1, Ure2, Swi1, Mot3, Cyc8, and Mod5) and one in Podospora anserina (HET-s). The article that reported the discovery of a prion form, the Mca1 protein, was retracted due to the fact that the data could not be reproduced. Notably, most of the fungal prions are based on glutamine/asparagine-rich sequences, with the exception of HET-s and Mod5.
Research into fungal prions has given strong support to the protein-only concept, since purified protein extracted from cells with a prion state has been demonstrated to convert the normal form of the protein into a misfolded form in vitro, and in the process, preserve the information corresponding to different strains of the prion state. It has also shed some light on prion domains, which are regions in a protein that promote the conversion into a prion. Fungal prions have helped to suggest mechanisms of conversion that may apply to all prions, though fungal prions appear distinct from infectious mammalian prions in the lack of cofactor required for propagation. The characteristic prion domains may vary between species—e.g., characteristic fungal prion domains are not found in mammalian prions.
Potential treatments and diagnosis
Advancements in computer modeling have allowed scientists to identify compounds that can treat prion-caused diseases, such as one compound found to bind a cavity in the PrPC and stabilize the conformation, reducing the amount of harmful PrPSc.
Antiprion antibodies capable of crossing the blood-brain-barrier and targeting cytosolic prion protein (an otherwise major obstacle in prion therapeutics) have been described.
In the last decade, some progress dealing with ultra-high-pressure inactivation of prion infectivity in processed meat has been reported.
In 2011, it was discovered that prions could be degraded by lichens.
There continues to be a very practical problem with diagnosis of prion diseases, including BSE and CJD. They have an incubation period of months to decades, during which there are no symptoms, even though the pathway of converting the normal brain PrP protein into the toxic, disease-related PrPSc form has started. At present, there is virtually no way to detect PrPSc reliably except by examining the brain using neuropathological and immunohistochemical methods after death. Accumulation of the abnormally folded PrPSc form of the PrP protein is a characteristic of the disease, but it is present at very low levels in easily accessible body fluids like blood or urine. Researchers have tried to develop methods to measure PrPSc, but there are still no fully accepted methods for use in materials such as blood.
In 2010, a team from New York described detection of PrPSc even when initially present at only one part in a hundred billion (10−11) in brain tissue. The method combines amplification with a novel technology called Surround Optical Fiber Immunoassay (SOFIA) and some specific antibodies against PrPSc. After amplifying and then concentrating any PrPSc, the samples are labelled with a fluorescent dye using an antibody for specificity and then finally loaded into a micro-capillary tube. This tube is placed in a specially constructed apparatus so that it is totally surrounded by optical fibres to capture all light emitted once the dye is excited using a laser.
The RT-QuIC assay, a microplate reader-based prion detection method which uses as reagents normally folded prions, fluorescently labelled so that they "light up" when they are misfolded; samples suspected of containing misfolded prions are added and misfolded reagents can be detected by standard fluorescence detection methods.
Astemizole has been found to have anti-prion activity.
Another type of chemical that may be effective against prion infection is the luminescent conjugated polythiophenes, fluorescent compounds that are often used to stain tissue samples. In a 2015 study, led by Adriano Aguzzi, professor of neurobiology at the University of Zurich, found that when they injected mice with a prion disease and then with polythiophenes, the mice survived eighty percent longer than the control mice that were only injected with the prion disease.
Causal hypotheses
Whether prions cause disease or are merely a symptom caused by a different agent is still debated by a minority of researchers. The following sections describe several hypotheses: Some pertain to the composition of the infectious agent (protein-only, protein with other components, virus, or other), while others pertain to its mechanism of reproduction.
Protein-only hypothesis
Prior to the discovery of prions, it was thought that all pathogens used nucleic acids to direct their replication. The "protein-only hypothesis" states that a protein structure can replicate without the use of nucleic acids. This was initially controversial as it contradicts the central dogma of molecular biology, which describes nucleic acid as the central form of replicative information.
Evidence in favor of a protein-only hypothesis includes:
Genetic factors
A gene for the normal protein has been identified: the PRNP gene. In all inherited cases of prion disease, there is a mutation in the PRNP gene. Many different PRNP mutations have been identified and these proteins are more likely to fold into abnormal prion. Although this discovery puts a hole in the general prion hypothesis, that prions can aggregate only proteins of identical amino acid make-up. These mutations can occur throughout the gene. Some mutations involve expansion of the octapeptide repeat region at the N-terminal of PrP. Other mutations that have been identified as a cause of inherited prion disease occur at positions 102, 117 & 198 (GSS), 178, 200, 210 & 232 (CJD) and 178 (Fatal Familial Insomnia, FFI). The cause of prion disease can be sporadic, genetic, or infectious, or a combination of these factors. For example, to have scrapie, both an infectious agent and a susceptible genotype must be present.
Multi-component hypothesis
Despite much effort, significant titers of prion infectivity have never been produced by refolding pure PrP molecules, raising doubt about the validity of the "protein only" hypothesis. In addition, the "protein only" hypothesis fails to provide a molecular explanation for the ability of prion strains to target specific areas of the brain in distinct patterns. These shortcomings, along with additional experimental data, have given rise to the "multi-component" or "cofactor variation" hypothesis.
In 2007, biochemist Surachai Supattapone and his colleagues at Dartmouth College produced purified infectious prions de novo from defined components (PrPC, co-purified lipids, and a synthetic polyanionic molecule). These researchers also showed that the polyanionic molecule required for prion formation was selectively incorporated into high-affinity complexes with PrP molecules, leading them to hypothesize that infectious prions may be composed of multiple host components, including PrP, lipid, and polyanionic molecules, rather than PrPSc alone.
In 2010, Jiyan Ma and colleagues at The Ohio State University produced infectious prions from a recipe of bacterially expressed recombinant PrP, POPG phospholipid, and RNA, further supporting the multi-component hypothesis. This finding is in contrast to studies that found minimally infectious prions produced from recombinant PrP alone.
In 2012, Supattapone and colleagues purified the membrane lipid phosphatidylethanolamine as a solitary endogenous cofactor capable of facilitating the formation of high-titer recombinant prions derived from multiple prion strains. They also reported that the cofactor is essential for maintaining the infectious conformation of PrPSc, and that cofactor molecules dictate the strain properties of infectious prions.
Heavy metal poisoning hypothesis
Reports suggest that imbalance of brain metal homeostasis may be a cause of PrPSc-associated neurotoxicity, though the underlying mechanisms are difficult to explain based on existing information. Proposed hypotheses include a functional role for PrPC in metal metabolism, and loss of this function due to aggregation to the disease-associated PrPSc form as the cause of brain metal imbalance. Other views suggest gain of toxic function by PrPSc due to sequestration of PrPC-associated metals within the aggregates, resulting in the generation of redox-active PrPSc complexes. The physiological implications of some PrPC-metal interactions are known, while others are still unclear. The pathological implications of PrPC-metal interaction include metal-induced oxidative damage, and in some instances conversion of PrPC to a PrPSc-like form.
Viral hypothesis
The protein-only hypothesis has been criticised by those maintaining that the simplest explanation of the evidence to date is viral. For more than a decade, Yale University neuropathologist Laura Manuelidis has been proposing that prion diseases are caused instead by an unidentified slow virus. In January 2007, she and her colleagues published an article reporting to have found a virus in 10%, or less, of their scrapie-infected cells in culture.
Evidence in favor of a viral hypothesis includes:
Studies propagating TSE infectivity in cell-free reactions and in purified component chemical reactions is thought to strongly suggest against TSE viral nature. However, some viruses, such as Poliovirus, have the ability to replicate in cell-free reactions.
Virino hypothesis
The 'virino hypothesis' postulates that the TSE agent is a foreign, self replicating nucleic acid or nucleic acid fragment bound to PrP.
Spiroplasma hypothesis
Spiroplasma is a cell wall–deficient bacteria related to Mycoplasma, which some think may be the cause of the TSEs. The lack of a cell wall means they are not susceptible to conventional antibiotics such as penicillin, which target cell wall synthesis. Frank O. Bastian of Louisiana State University first discovered Spiroplasma-like inclusions in the brain of a CJD patient during an autopsy in 1979 and has hypothesized that this bacterium could possibly be the cause of the TSEs.
However, as of 2015, with the exception of Spiroplasma mirum strain SMCA causing spongiform microcystic encephalitis in suckling rats, other researchers have been unable to duplicate these findings, casting doubt on the Spiroplasma hypothesis. In defense of the Spiroplasma hypothesis, Bastian pointed out that Spiroplasma is hard to culture and that strain variation makes it hard to detect certain strains using PCR and other techniques, thus giving a false negative.
Acinetobacter-autoimmunity hypothesis
Acinetobacter is a bacterium which some think is the cause of the TSEs.
Etymology and pronunciation
The word prion, coined in 1982 by Stanley B. Prusiner, is a portmanteau derived from protein and infection, hence prion, and is short for "proteinaceous infectious particle", in reference to its ability to self-propagate and transmit its conformation to other proteins. Its main pronunciation is /ˈpriːɒn/, although /ˈpraɪɒn/, as the homographic name of the bird is pronounced, is also heard.