
 | ||
The Payne rearrangement is the isomerization, under basic conditions, of 2,3-epoxy alcohols to isomeric 2,3-epoxy alcohols with inversion of configuration. Aza- and thia-Payne rearrangements of aziridines and thiiraniums, respectively, are also known.
Contents
Introduction
Under basic, protic conditions, 2,3-epoxy alcohols undergo a rearrangement in which the alcohol oxygen opens the epoxide with inversion of configuration, forming an isomeric 1,2-epoxy alcohol. Overall, the Payne rearrangement represents a migration of the epoxide. Although the migration itself is fully reversible, nucleophilic opening under Curtin–Hammett conditions provides good yields of functionalized diols derived from a single epoxy alcohol isomer. Intramolecular electrophilic trapping of the new alkoxide generated upon rearrangement may also be used to drive the reaction to completion. In some cases, the thermodynamic difference between epoxide isomers is large enough to obtain a single isomer in synthetically useful yield without relying on kinetic differences associated with trapping.
(1)
Strongly basic conditions are required to induce equilibration, which limits the synthetic utility of the transformation to substrates lacking base-labile functionality. Many epoxy alcohol equilibria are very finely balanced; however, taking advantage of the trapping strategies described above may lead to high yields of single isomers.
Prevailing mechanism
The basic mechanism of the Payne rearrangement involves deprotonation of the free hydroxyl group, invertive nucleophilic attack on the proximal epoxide carbon, and re-protonation of the newly freed alkoxide. Each step of the process is reversible.
(2)
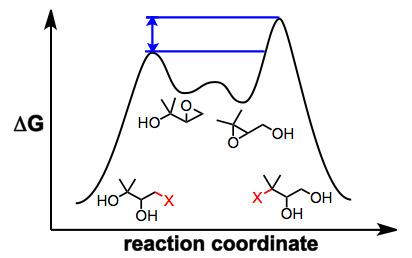
Several observations suggest that this mechanistic picture is oversimplified. Epoxide migration either does not occur or is very sluggish under aprotic conditions—it has been suggested that nucleophilic attack is slowed by the coordination of metal ions to the nucleophilic oxygen under aprotic conditions. In addition, when an external nucleophile is added to equilibrating epoxide isomers, the ratio of opened products does not reflect the ratio of epoxide isomers in solution or their relative thermodynamic stability. In situ nucleophilic opening of equilibrating epoxides is an example of Curtin-Hammett conditions—because the epoxides are equilibrating rapidly relative to the rate of epoxide opening, it is the kinetic barriers of ring opening that control the observed product ratio. In the example below, the product of opening of the terminal epoxide is the major product, even though the terminal epoxide itself is less thermodynamically stable than the internal isomer.
(3)
Halo diols may be used as precursors to 2,3-epoxy alcohols prior to rearrangement. Issues of site selectivity may arise if the two hydroxyl groups flanking the halide are not equivalent. In general, the formation of internal, substituted epoxides is more rapid than the formation of terminal epoxides. This idea can be used to predict the course of migrations of in situ-generated epoxides.
(4)
Stereochemistry
The Payne rearrangement occurs with inversion of stereochemistry at C-2. Substrates containing multiple adjacent hydroxyl groups may undergo "cascade" epoxide migrations with inversion at each site of nucleophilic attack. In one example, inversion of three contiguous stereocenters results after two epoxide migrations, opening of the epoxide by carboxylate, and hydrolysis of the resulting lactone.
(5)
Payne rearrangement
The position of equilibrium in both cyclic and acyclic systems may be predicted from the structures of the two equilibrating epoxides. In acyclic systems, these rules have been established:
Pyranosides are the most heavily studied cyclic systems. Studies of epoxide migration in pyranosides and other cyclic epoxy alcohols have revealed three generalizations:
Conformationally locked pyranosides reveal the thermodynamic preference of cyclic substrates for more pseudoequatorial groups.
(6)
Under aprotic conditions, nucleophilic opening of epoxide isomers can be achieved with hydrides or organocuprates. Nucleophilic attack generally takes place at the least substituted carbon, yielding the more substituted diol product.
(7)
Under protic conditions, opening at the least substituted position is also usually favored. Nucleophiles that may be used under protic conditions include phenols, secondary amines, azide anion, and sulfides.
(8)
Intermolecular nucleophilic trapping of a single epoxide isomer is difficult, as reaction of the epoxy alcohol with the electrophile is typically faster than migration. However, intramolecular electrophies are often effective for trapping a single epoxide isomer. For instance, a second nearby epoxide in the starting material of equation (9) is trapped by a single epoxide isomer, leading to a tetrahydrofuran.
(9)
Aza- and thia-payne rearrangements
The aza-Payne rearrangement may be effected in either the "forward" (epoxide to aziridine) or "reverse" (aziridine to epoxide) direction depending on the conditions employed. Electron-poor aziridines undergo the reverse rearrangement in the presence of hydride base, while the corresponding epoxy amines undergo the forward rearrangement in the presence of boron trifluoride etherate.
(10)
The thia-Payne rearrangement has only been observed in the forward direction (epoxide to thiiranium) with in situ opening of the thiiranium. Invertive nucleophilic opening at C-2 is possible through the use of trialkylaluminum reagents.
(11)
Synthetic applications
The synthesis of borjatriol involved the rare isolation of a migrated epoxide. The diastereomeric mixture of rearrangement products was carried through the remainder of the synthesis.
(12)
The final two steps in the total synthesis of spatol involved intramolecular electrophilic trapping of an alkoxide derived from a rearranged epoxide. Attack of the intermediate alkoxide on the adjacent mesylate afforded a bis(epoxide), and debenzylation provided the target compound.
(13)
Comparison with other methods
Other methods available for the preparation of 2,3-epoxy alcohols have the advantage that they do not begin with an existing 2,3-epoxy alcohol; however, they tend to involve more steps than epoxide migration. Asymmetric dihydroxylation may be used to synthesize epoxy alcohols with high stereoselectivity, and some of the methods relying on dihydroxylation avoid the use of strongly basic conditions.
(14)
An alternative method that leads to retention of configuration at C-2 involves mesylation of an epoxy alcohol, epoxide opening, and re-closing by displacement of the mesylate.
(15)
Typical conditions
Opening of terminal epoxides by adventitious hydroxide may occur under the conditions of rearrangement; if this is not desired, anhydrous solvents, reagents, and glassware must be used. Freshly prepared sodium methoxide in methanol is commonly used to effect rearrangement without opening. Nucleophilic opening can be accomplished through the use of sodium azide, excess hydroxide, or cuprate reagents in the presence of lithium chloride. Electrophilic trapping is carried out under standard conditions in the presence of an electrophile such as benzyl bromide. Silyl halides have also been used as electrophilic trapping agents.
To prevent epoxide migration, weakly basic conditions may be employed. Neither aqueous potassium carbonate nor aqueous amine bases cause epoxide rearrangement. Low temperatures are also beneficial when epoxide migration is not desired.
Example procedure
(16)
A solution of methyl(cyano)cuprate (Solution A) was prepared as follows: to a suspension of 0.35 g (3.91 mmol) of copper(I) cyanide in 5 mL of tetrahydrofuran under argon at 0° was added dropwise over about 5 minutes 2.76 mL of a solution of methyllithium in ethyl ether (1.4 M, 3.86 mmol). The colorless solution was stirred for 10 minutes at 0°, warmed to 25° over 30 minutes, then cooled again to 0°. Separately, a solution of the lithium salt of (±)-cis-4-benzyloxy-2,3-epoxy-1-butanol (Solution B) was prepared as follows: to a solution of 0.5 g (2.58 mmol) of the epoxy alcohol and 0.90 g (21.4 mmol) of lithium chloride in 10 mL of tetrahydrofuran under argon at −78° was added dropwise 1.65 mL of a solution of n-butyllithium in hexane (1.56 M, 2.58 mmol). The solution was stirred for 5 minutes at −78°, allowed to warm to 0°, and then stirred at that temperature for 10 minutes. The reaction was effected by the addition of Solution A to Solution B via cannula at 0° followed by warming to room temperature over 2 hours. The reaction mixture was then stirred for a further 12 hours and then cautiously treated with 5 mL of saturated aqueous ammonium chloride. The mixture was stirred for 1–2 hours to aid removal of copper residues. Ethyl ether (20 mL) was then added, and the organic layer was separated. The aqueous phase was extracted twice with 20 mL of ethyl ether, and the combined organic phases were dried over magnesium sulfate, filtered, and concentrated to give 0.51 g of the product as a colorless oil (95%), IR (film) 3400, 3100, 3060, 3030, 2970, 2930, 2870, 1600, 1500, 1465, 1445, 1385, 1370, 1320, 1285, 1210, 1180, 1120, 1100, 1075, 1030, 1020, 980, 905, 830, 750, 730, 710, 695 cm–1; 1H NMR (CDCl3) δ 0.90 (t, J = 6.0 Hz, 3 H), 1.37–1.53 (m, 2 H), 3.20 (br s, 2 H), 3.40–3.65 (m, 4 H), 4.48 (s, 2 H), 7.29 (s, 5 H).