
Specialty urology OMIM 609814 305800 MedlinePlus 000475 | ICD-9-CM 581.2, 582.2, 583.2 DiseasesDB 34457 | |
 | ||
ICD-10 N00-N08 with .5 and .6 suffix | ||
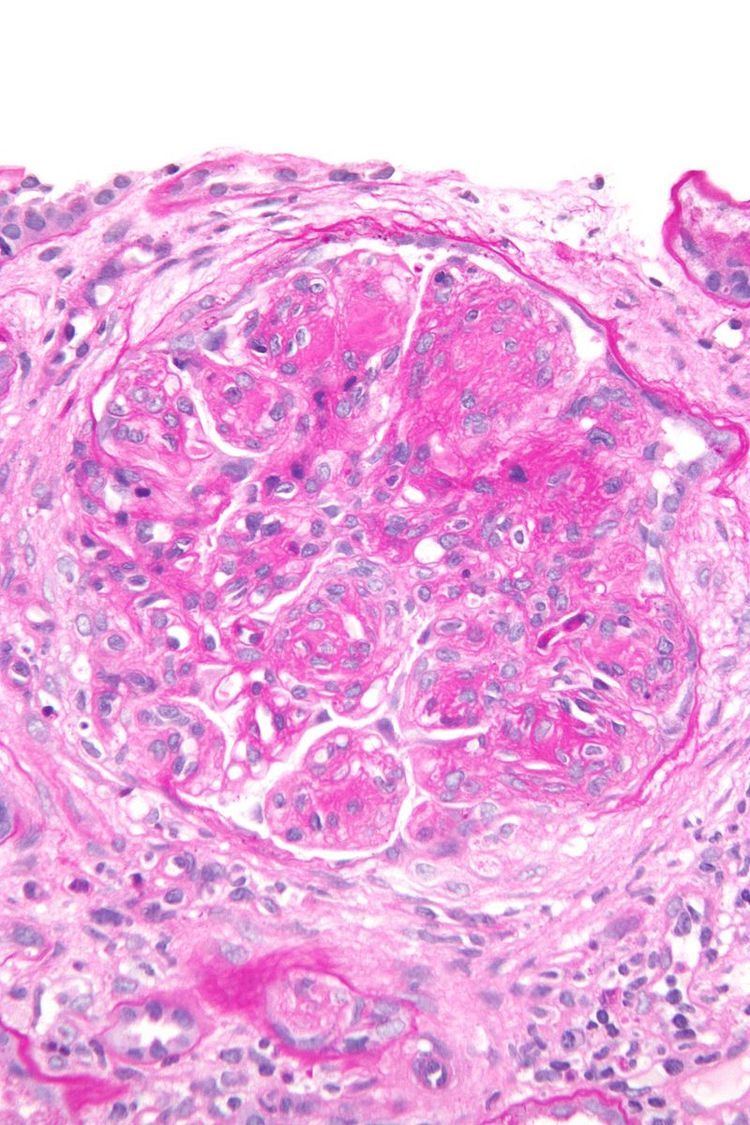
Membranoproliferative glomerulonephritis ("MPGN"), also known as mesangiocapillary glomerulonephritis, is a type of glomerulonephritis caused by deposits in the kidney glomerular mesangium and basement membrane (GBM) thickening, activating complement and damaging the glomeruli.
Contents
MPGN accounts for approximately 4% of primary renal causes of nephritic syndrome in children and 7% in adults.
It should not be confused with membranous glomerulonephritis, a condition in which the basement membrane is thickened, but the mesangium is not.
Pathology
Membranoproliferative glomerulonephritis involves deposits at the intraglomerular mesangium.
It is also the main hepatitis C associated nephropathy.
The histomorphologic differential diagnosis includes transplant glomerulopathy and thrombotic microangiopathies.
Appearance
The GBM is rebuilt on top of the deposits, causing a "tram tracking" appearance under the microscope. Mesangial cellularity is increased.
Type
There are three types of MPGN, but this classification is becoming obsolete as the causes of this pattern are becoming understood.
Type I
Type I the most common by far, is caused by immune complexes depositing in the kidney. It is characterised by subendothelial and mesangial immune deposits.
It is believed to be associated with the classical complement pathway.
Type II
The preferred name is "dense deposit disease". Most cases of dense deposit disease do not show a membranoproliferative pattern, A 2012 review considers DDD to be in a continuum with C3 glomerulonephritis, one reason the use of type I to type III classification system is falling out of favour.
Most cases are associated with the dysregulation of the alternative complement pathway.
Spontaneous remissions of MPGN II are rare, approximately half of those affected with MPGN II will progress to end stage renal disease within 10 years.
In many cases, people with MPGN II can develop drusen which is caused by same deposits within the Bruch's membrane beneath the retinal pigment epithelium (RPE) layer of the eye. Over time vision can deteriorate and subretinal neovascular membranes, macular detachment, and central serous retinopathy develop.
Type III
Type III is very rare, it is characterized by a mixture of subepithelial deposits and the typical pathological findings of Type I disease.
A candidate gene has been identified on chromosome 1.
Complement component 3 is seen under immunofluorescence.