
Development status Active Operating system | Type plugin | |
 | ||
Developer(s) Insilab (National Institute of Chemistry Slovenia) Initial release 2015; 2 years ago (2015) | ||
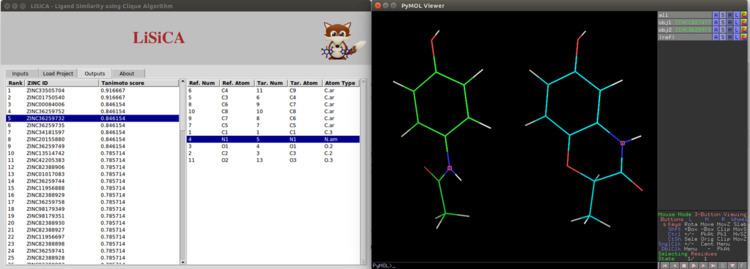
LiSiCA (Ligand Similarity using Clique Algorithm) is a ligand-based virtual screening software that searches for 2D and 3D similarities between a reference compound and a database of target compounds which should be represented in a Mol2 format. The similarities are expressed using the Tanimoto coefficients and the target compounds are ranked accordingly. LiSiCA is also available as LiSiCA PyMOL plugin both on Linux and Windows operating systems.
Contents
Description
As an input LiSiCA requires at least one reference compound and database of target compounds. For 3D screening this database has to be a pregenerated database of conformations of target and for 2D screening a topology, that is, a list of atoms and bonds, for each target compound. On each step the algorithm compares reference compound to one of the compounds from target compounds based on their 2D or 3D representation. Both compounds(molecules) are converted to molecular graphs. In 2D and 3D screening the molecular graph vertices represent atoms. In 2D screening edges of molecular graph represent covalent bonds while in 3D screening edges are drawn between every pair of vertices and have no chemical meaning. A product graph generated from molecular graphs is then searched using fast maximum clique algorithm to find the largest substructure common to both compounds. The similarity between compounds is calculated using Tanimoto coefficients and target compounds are ranked according to their Tanimoto coefficients.
Feature overview
LiSiCA can search 2D and 3D similarities between a reference compound and a database of target compounds. It takes as an input at least one reference compound and a database of target compounds. By default it returns only the compound most similar to the reference compound out of all compounds in database of target compounds. Other optional parameters LiSiCA uses are:
Default value: the default is to try to detect the number of CPUs and use all of them or, failing that, use 1
Possible input: 2, 3
Default value: 2
Default value: 1.0
Default value: 1
Default value: False
Default value: 0
Default value: 1
In addition LiSiCA PyMOL plugin also offers to load saved results.
History
Interesting fact
The word lisica in Slovenian language means a fox that is why the logo of LiSiCA software is a fox holding two molecules.