
EC number 3.2.1.52 IntEnz IntEnz view ExPASy NiceZyme view | CAS number 9012-33-3 BRENDA BRENDA entry KEGG KEGG entry | |
 | ||
Hexosaminidase (EC 3.2.1.52, beta-acetylaminodeoxyhexosidase, N-acetyl-beta-D-hexosaminidase, N-acetyl-beta-hexosaminidase, N-acetyl hexosaminidase, beta-hexosaminidase, beta-acetylhexosaminidinase, beta-D-N-acetylhexosaminidase, beta-N-acetyl-D-hexosaminidase, beta-N-acetylglucosaminidase, hexosaminidase A, N-acetylhexosaminidase, beta-D-hexosaminidase) is an enzyme involved in the hydrolysis of terminal N-acetyl-D-hexosamine residues in N-acetyl-β-D-hexosaminides.
Contents
Lysosomal A, B, and S isozymes
Functional lysosomal β-hexosaminidase enzymes are dimeric in structure. Three isozymes are produced through the combination of α and β subunits to form any one of three active dimers:
The α and β subunits are encoded by separate genes, HEXA and HEXB respectively. Beta-hexosaminidase and the cofactor GM2 activator protein catalyze the degradation of the GM2 gangliosides and other molecules containing terminal N-acetyl hexosamines. Gene mutations in HEXB often result in Sandhoff disease; whereas, mutations in HEXA decrease the hydrolysis of GM2 gangliosides, which is the main cause of Tay-Sachs disease.
Function
Even though the alpha and beta subunits of lysosomal hexosaminidase can both cleave GalNAc residues, only the alpha subunit is able to hydrolyze GM2 gangliosides because of a key residue, Arg-424, and a loop structure that forms from the amino acid sequence in the alpha subunit. The loop in the alpha subunit, consisting of Gly-280, Ser-281, Glu-282, and Pro-283 which is absent in the beta subunit, serves as an ideal structure for the binding of the GM2 activator protein (GM2AP), and arginine is essential for binding the N-acetyl-neuraminic acid residue of GM2 gangliosides. The GM2 activator protein transports GM2 gangliosides and presents the lipids to hexosaminidase, so a functional hexosaminidase enzyme is able to hydrolyze GM2 gangliosides into GM3 gangliosides by removing the N-acetylgalactosamine (GalNAc) residue from GM2 gangliosides.
Mechanism of action
A Michaelis complex consisting of a glutamate residue, a GalNAc residue on the GM2 ganglioside, and an aspartate residue leads to the formation of an oxazolinium ion intermediate. A glutamate residue (alpha Glu-323/beta Glu-355) works as an acid by donating its hydrogen to the glycosidic oxygen atom on the GalNAc residue. An aspartate residue (alpha Asp-322/beta Asp-354) positions the C2-acetamindo group so that it can be attacked by the nucleophile (N-acetamido oxygen atom on carbon 1 of the substrate). The aspartate residue stabilizes the positive charge on the nitrogen atom in the oxazolinium ion intermediate. Following the formation of the oxazolinium ion intermediate, water attacks the electrophillic acetal carbon. Glutamate acts as a base by deprotonating the water leading to the formation of the product complex and the GM3ganglioside.
Gene mutations resulting in Tay-Sachs Disease
There are numerous mutations that lead to hexosaminidase deficiency including gene deletions, nonsense mutations, and missense mutations. Tay-Sachs disease occurs when hexosaminidase A loses its ability to function. People with Tay-Sachs disease are unable to remove the GalNAc residue from the GM2 ganglioside, and as a result, they end up storing 100 to 1000 times more GM2 gangliosides in the brain than the normal person. Over 100 different mutations have been discovered just in infantile cases of Tay-Sachs disease alone.
The most common mutation, which occurs in over 80 percent of Tay-Sachs patients, results from a four base pair addition (TATC) in exon 11 of the Hex A gene. This insertion leads to an early stop codon, which causes the Hex A deficiency.
Children born with Tay-Sachs usually die between two and four years of age from aspiration and pneumonia. Tay-Sachs causes cerebral degeneration and blindness. Patients also experience flaccid extremities and seizures. At this point in time, there has been no cure or effective treatment of Tay-Sachs disease.
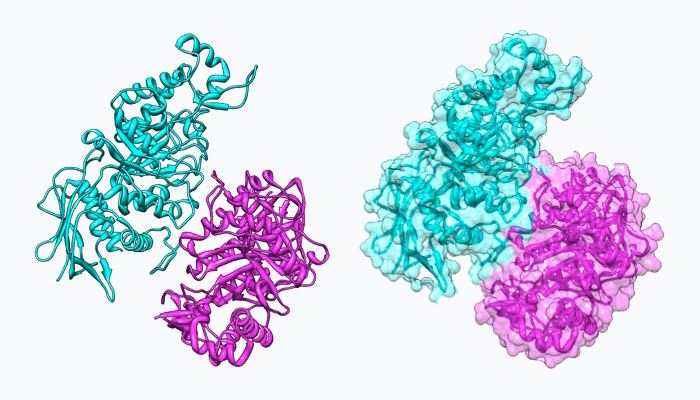
NAG-thiazoline, NGT, acts as mechanism based inhibitor of hexosaminidase A. In patients with Tay-Sachs disease (misfolded hexosaminidase A), NGT acts as a molecular chaperone by binding in the active site of hexosaminidase A which helps create a properly folded hexosaminidase A. The stable dimer conformation of hexosaminidase A has the ability to leave the endoplasmic reticulum and is directed to the lysosome where it can perform the degradation of GM2 gangliosides. The two subunits of hexosaminidase A are shown below:
Cytosolic C and D isozymes
The bifunctional protein NCOAT (nuclear cytoplasmic O-GlcNAcase and acetyltransferase) that is encoded by the MGEA5 gene possesses both hexosaminidase and histone acetyltransferase activities. NCOAT is also known as hexosaminidase C and has distinct substrate specificities compared to lysosomal hexosaminidase A. A single-nucleotide polymorphism in the human O-GlcNAcase gene is linked to diabetes mellitus type 2.
A fourth mammalian hexosaminidase polypeptide which has been designated hexosaminidase D (HEXDC) has recently been identified.