
Species Human Entrez 3043 | Human Mouse Ensembl ENSG00000244734 | |
 | ||
Aliases HBB, CD113t-C, beta-globin, hemoglobin subunit beta External IDs MGI: 5474852 HomoloGene: 68066 GeneCards: HBB | ||
Beta globin (also referred to as HBB, β-globin, haemoglobin beta, hemoglobin beta, or preferably haemoglobin subunit beta) is a globin protein, which along with alpha globin (HBA), makes up the most common form of haemoglobin in adult humans, the HbA. It is 146 amino acids long and has a molecular weight of 15,867 Da. Normal adult human HbA is a heterotetramer consisting of two alpha chains and two beta chains.
Contents
- Gene locus
- Interactions
- Beta thalassemia
- Sickle cell disease
- Haemoglobin C
- Haemoglobin E
- Human evolution
- References
HBB is encoded by the HBB gene on human chromosome 11. Mutations in the gene produce several variants of the proteins which are implicated with genetic disorders such as sickle-cell disease and beta thalassemia, as well as beneficial traits such as genetic resistance to malaria.
Gene locus
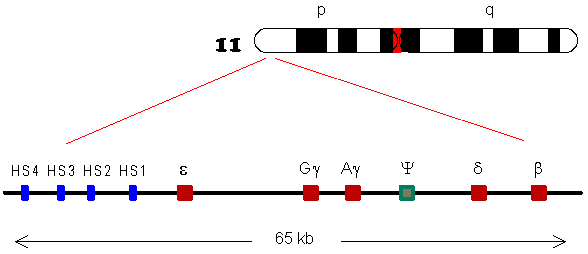
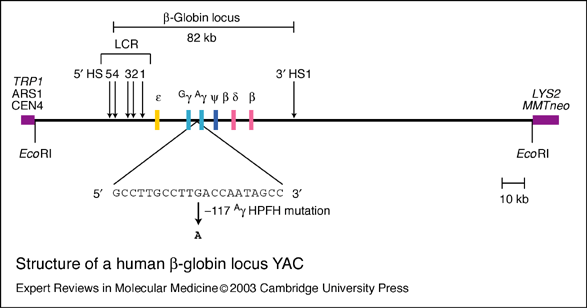
HBB protein is produced by the gene HBB which is located in the multigene locus of β-globin locus on chromosome 11, specifically on the short arm position 15.5. Expression of beta globin and the neighbouring globins in the β-globin locus is controlled by single locus control region (LCR), the most important regulatory element in the locus located upstream of the globin genes. The normal allelic variant is 1600 base pairs (bp) long and contains three exons. The order of the genes in the beta-globin cluster is 5' - epsilon – gamma-G – gamma-A – delta – beta - 3'.
Interactions
HBB interacts with Hemoglobin, alpha 1 (HBA1) to form haemoglobin A, the major haemoglobin in adult humans. The interaction is two-fold. First, one HBB and one HBA1 combine, non-covalently, to form a dimer. Secondly, two dimers combine to form the four-chain tetramer, and this becomes the functional haemolglobin.
Beta thalassemia
Total or partial absence of HBB causes a genetic disease called beta thalassemia. Total loss called, thalassemia major or beta-0-thalassemia, is due to mutation in both alleles, and this results in failure to form beta chain of haemoglobin. It prevents oxygen supply in the tissues. It is highly lethal. Symptoms, such as severe anaemia and heart attack, appear within two years after birth. They can be treated only by lifelong blood transfusion and bone marrow transplantation. Reduced HBB function called thalassemia minor or beta+ thalassemia is due to mutation in one of the alleles. It is less severe but patients are prone to other diseases such as asthma and liver problems.
According to a recent study, the stop gain mutation Gln40stop in HBB gene is a common cause of autosomal recessive Beta- thalassemia in Sardinian people(almost exclusive in Sardinia). Carriers of this mutation show an enhaced red blood cell count. As a curiosity, the same mutation was also associated to a decrease in serum LDL levels in carriers, so the authors suggest that is due to the need of cholesterol to regenerate cell membranes.
Sickle cell disease
More than a thousand naturally occurring HBB variants have been discovered. The most common is HbS, which causes sickle cell disease. HbS is produced by a point mutation in HBB in which the codon GAG is replaced by GTG. This results in the replacement of hydrophilic amino acid glutamic acid with the hydrophobic amino acid valine at the sixth position (β6Glu→Val). This substitution creates a hydrophobic spot on the outside of the protein that sticks to the hydrophobic region of an adjacent haemoglobin molecule's beta chain. This further causes clumping of HbS molecules into rigid fibers, causing "sickling" of the entire red blood cells in the homozygous (HbS/HbS) condition. Homozygous allele has become one of the deadliest genetic factors. Whereas, people heterozygous for the mutant allele (HbS/HbA) are resistant to malaria and develop minimal effects of the anaemia.
Haemoglobin C
Sickle cell disease is closely related to another mutant haemoglobin called haemoglobin C (HbC), because they can be inherited together. HbC mutation is at the same position in HbS, but glutamic acid is replaced by lysine (β6Glu→Lys). The mutation is particularly prevalent in West African populations. HbC provides near full protection against Plasmodium falciparum in homozygous (CC) individuals and intermediate protection in heterozygous (AC) individuals. This indicates that HbC has stronger influence than HbS, and is predicted to replace HbS in malaria-endemic regions.
Haemoglobin E
Another point mutation in HBB, in which glutamic acid is replaced with lysine at position 26 (β26Glu→Lys), leads to the formation of haemoglobin E (HbE). HbE has a very unstable α- and β-globin association. Even though the unstable protein itself has mild effect, inherited with HbS and thalassemia traits, it turns into a life-threatening form of β-thalassemia. The mutation is of relatively recent origin suggesting that it resulted from selective pressure against severe falciparum malaria, as heterozygous allele prevents the development of malaria.
Human evolution
Malaria due to Plasmodium falciparum is a major selective factor in human evolution. It has influenced mutations in HBB in various degrees resulting in the existence of numerous HBB variants. Some of these mutations are not directly lethal and instead confer resistance to malaria, particularly in Africa where malaria is epidemic. People of African descent have evolved to have higher rates of the mutant HBB because the heterozygous individuals have a misshaped red blood cell that prevent attacks from malarial parasites. Thus, HBB mutants are the sources of positive selection in these regions and are important for their long-term survival. Such selection markers are important for tracing human ancestry and diversification from Africa.