
EC number 3.1.3.9 ExPASy NiceZyme view | CAS number 9001-39-2 | |
 | ||
Glucose 6-phosphatase (EC 3.1.3.9, G6Pase) is an enzyme that hydrolyzes Glucose-6-phosphate, resulting in the creation of a phosphate group and free glucose. Glucose is then exported from the cell via glucose transporter membrane proteins. This catalysis completes the final step in gluconeogenesis and glycogenolysis and therefore plays a key role in the homeostatic regulation of blood glucose levels.
Contents
- Glucose 6 phosphate Dehydrogenase G6PD Enzyme
- Structure and function
- Mechanism
- Expression
- Clinical significance
- References
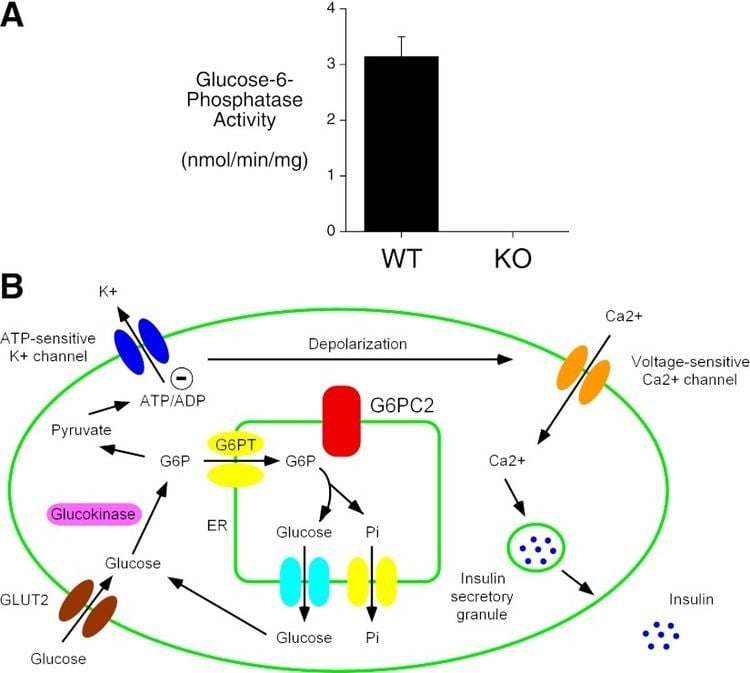
Glucose-6-phosphatase is a complex of multiple component proteins, including transporters for G6P, glucose, and phosphate. The main phosphatase function is performed by the glucose-6-phosphatase catalytic subunit. In humans, there are three isozymes of the catalytic subunit: glucose-6-phosphatase-α, encoded by G6PC; IGRP, encoded by G6PC2; and glucose-6-phosphatase-β, encoded by G6PC3.
Glucose-6-phosphatase-α and glucose-6-phosphatase-β are both functional phosphohydrolases, and have similar active site structure, topology, mechanism of action, and kinetic properties with respect to G6P hydrolysis. In contrast, IGRP has almost no hydrolase activity, and may play a different role in stimulating pancreatic insulin secretion.
![Cellular Release of [18F]2-Fluoro-2-deoxyglucose as a Function of the Glucose-6-phosphatase Enzyme System* - Journal of Biological Chemistry](https://alchetron.com/cdn/glucose-6-phosphatase-b568376b-33ac-4312-92a0-6acfad04a0a-resize-750.jpg)
Glucose-6-phosphate Dehydrogenase (G6PD) Enzyme
Structure and function
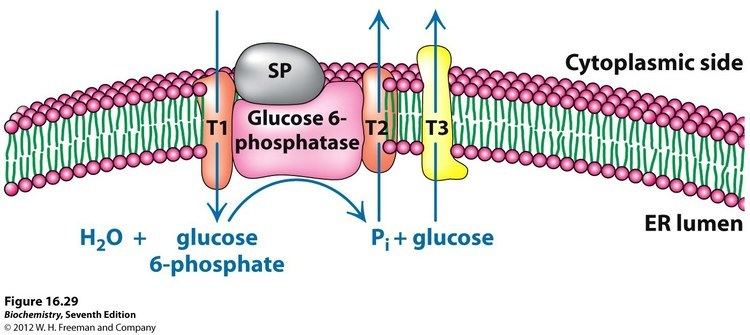
Although a clear consensus has not been reached, a large number of scientists adhere to a substrate-transport model to account for the catalytic properties of glucose-6-phosphatase. In this model, glucose-6-phosphatase has a low degree of selectivity. The transfer of the glucose 6-phosphate is carried out by a transporter protein (T1) and the endoplasmic reticulum (ER) contains structures allowing the exit of the phosphate group (T2) and glucose (T3).
Glucose-6-phosphatase consists of 357 amino acids, and is anchored to the endoplasmic reticulum (ER) by nine transmembrane helicies. Its N-terminal and active site are found on the lumen side of the ER and its C-terminus projects into the cytoplasm. Due to its tight association to the ER, the exact structure of glucose-6-phosphatase remains unknown. However, sequence alignment has shown that glucose-6-phosphatase is structurally similar to the active site of the vanadium-containing chloroperoxidase found in Curvularia inaequalis.
Based on pH kinetic studies of glucose-6-phosphatase-α catalysis, it was proposed that the hydrolysis of glucose-6-Phosphate was completed via a covalent phosphohistidine glucose-6-Phosphate intermediate. The active site of glucose-6-phosphatase-α was initially identified by the presence of a conserved phosphate signature motif usually found in lipid phosphatases, acid phosphatases, and vanadium haloperoxidases.
Essential residues in the active site of vanadium haloperoxidases include: Lys353, Arg360, Arg490, His404, and His496. Corresponding residues in the active site of glucose-6-phosphatase-α include Arg170 and Arg83, which donate hydrogen ions to the phosphate, stabilizing the transition state, His119, which provides a proton to the dephosphorylated oxygen attached to glucose, and His176, which completes a nucleophilic attack on the phosphate to form a covalently bound phosphoryl enzyme intermediate. Within the Vanadium-containing chloroperoxidase, Lys353 was found to stabilize the phosphate in the transition state. However, the corresponding residue in glucose-6-phosphatase-α (Lys76) resides within the ER membrane and its function, if any, is currently undetermined. With the exception of Lys76, these residues are all located on the luminal side of the ER membrane.
Glucose-6-phosphatase-β is a ubiquitously expressed, 346-amino acid membrane protein that shares 36% sequence identity with glucose-6-phosphatase-α. Within the glucose-6-phosphatase-β enzyme, sequence alignments predict that its active site contains His167, His114, and Arg79. Similar to that of the glucose-6-phosphatase-α active site, His167 is the residue that provides the nucleophilic attack, and His114, and Arg79 are the hydrogen donors. glucose-6-phosphatase-β is also localized in the ER membrane, although its orientation is unknown.
Mechanism
The hydrolysis of glucose-6-phosphate begins with a nucleophilic attack on the sugar-bound phosphate by His176 resulting in the formation of a phosphohistidine bond and the degradation of a carbonyl. A Negatively charged oxygen then transfers its electrons reforming a carbonyl and breaking its bond with glucose. The negatively charged glucose-bound oxygen is then protonated by His119 forming a free glucose. The phospho-intermediate produced by the reaction between His176 and the phosphate group is then broken by a hydrophilic attack; after the addition of another hydroxide and the decomposition of a carbonyl, the carbonyl is reformed kicking off the electrons originally donated by the His176 residue thereby creating a free phosphate group and completing the hydrolysis.
Expression
Genes coding for the enzyme are primarily expressed in the liver, in the kidney cortex and (to a lesser extent) in the β-cells of the pancreatic islets and intestinal mucosa (especially during times of starvation). According to Surholt and Newsholme, Glc 6-Pase is present in a wide variety of muscles across the animal kingdom, albeit at very low concentrations. Thus, the glycogen that muscles store is not usually available for the rest of the body's cells because glucose-6-phosphate cannot cross the sarcolemma unless it is dephosphorylated. The enzyme plays an important role during periods of fasting and when glucose levels are low. It has been shown that starvation and diabetes induces a 2-3-fold increase in glucose-6-phosphatase activity in the liver. Glc 6-Pase activity also increases dramatically at birth when an organism becomes independent of the mothers source of glucose. The human Glc 6-Pase gene contains five exons spanning approximately 125.5 kb DNA located on chromosome 17q21.
Clinical significance
Mutations of the glucose-6-phosphatase system, to be specific the glucose-6-phosphatase-α subunit (glucose-6-phosphatase-α), glucose-6-transporter (G6PT), and glucose-6-phosphatase-β (glucose-6-phosphatase-β or G6PC3) subunits lead to deficiencies in the maintenance of interprandial glucose homeostasis and neutrophil function and homeostasis. Mutations in both glucose-6-phosphatase-α and G6PT lead to Glycogen Storage Disease Type -1 (GSD-1) called von Gierke's disease. To be specific, mutations in the glucose-6-phosphatase-α lead to Glycogen Storage Disease Type-1a, which is characterized by accumulation of glycogen and fat in the liver and kidneys, resulting in hepatomegaly and renomegaly. GSD-1a constitutes approximately 80% of GSD-1 cases that present clinically. Absence of G6PT leads to GSD-1b (GSD-1b), which is characterized by the lack of a G6PT and represents 20% of the cases that present clinically.
The specific cause of the GSD-1a stems from nonsense mutations, insertions/deletions with or without a shift in the reading frame, or splice site mutations that occur at the genetic level. The missense mutations affect the two large luminal loops and transmembrane helices of glucose-6-phosphatase-α, abolishing or greatly reducing activity of the enzyme. The specific cause of GSD-1b stems from "severe" mutations such as splice site mutations, frame-shifting mutations, and substitutions of a highly conserved residue that completely destroyed G6PT activity. These mutations lead to the prevalence of GSD-1 by preventing the transport of glucose-6-phosphate (G6P) into the luminal portion of the ER and also inhibiting the conversion of G6P into glucose to be used by the cell.
The third type of glucose-6-phosphatase deficiency, glucose-6-phosphatase-β deficiency, is characterized by a congenital neutropenia syndrome in which neutrophils exhibit enhanced endoplasmic reticulum (ER) stress, increased apoptosis, impaired energy homeostasis, and impaired functionality. It can also lead to cardiac and urogenital malformations. This third class of deficiency is also affected by a G6PT deficiency as glucose-6-phosphatase-β also lies within the ER lumen and thus can lead to similar symptoms of glucose-6-phosphatase-β deficiency be associated with GSD-1b. Furthermore, recent studies have elucidated this area of similarity between both deficiencies and have shown that aberrant glycosylation occurs in both deficiencies. The neutrophil glycosylation has a profound effect on neutrophil activity and thus may also be classified as a congenital glycosylation disorder as well.
The major function of glucose-6-phosphatase-β has been determined to provide recycled glucose to the cytoplasm of neutrophils in order maintain normal function. Disruption of the glucose to G6P ratio due to significant decrease intracellular glucose levels cause significant disruption of glycolysis and HMS. Unless countered by uptake of extracellular glucose this deficiency leads to neutrophil dysfunction.
Interestingly, vanadium compounds such as vanadyl sulfate have been shown to inhibit the enzyme, and thus increase the insulin sensitivity in vivo in diabetics, as assessed by the hyperinsulinemic clamp technique, which may have potential therapeutic implications