
Specialty hematology ICD-9-CM 238.71 OMIM 187950 | ICD-10 D75.2, D47.3 ICD-O M9962/3 DiseasesDB 4522 | |
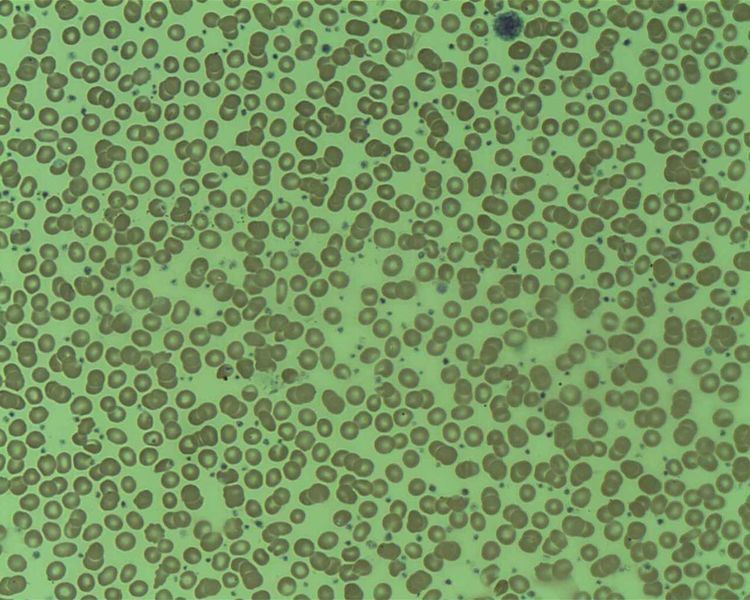 | ||
Essential thrombocytosis (ET; also known as essential thrombocythemia, essential thrombocythaemia, primary thrombocytosis) is a rare chronic blood disorder characterised by the overproduction of platelets by megakaryocytes in the bone marrow. It may, albeit rarely, develop into acute myeloid leukaemia or myelofibrosis. It is one of four myeloproliferative disorders (disorders characterised by increased production of a particular line of blood cell).
Contents
Signs and symptoms
Most people with ET are without symptoms referable to ET at the time of diagnosis, which is usually ultimately made after noting an elevated platelet level on a routine complete blood count (CBC). The most common symptoms are bleeding, blood clots, headache, nausea, vomiting, abdominal pain, visual disturbances, dizziness, fainting, and numbness in the extremities; the most common signs are increased white blood cell count, reduced red blood cell count, and enlarged spleen.
Cause
In ET, megakaryocytes are more sensitive to growth factors [citation needed]. Platelets derived from the abnormal megakaryocytes are activated, which, along with the elevated platelet count, contributes to the likelihood of thrombosis. The increased possibility of bleeding when the platelet count is over 1 million is due to von Willebrand factor (vWF) sequestration by the increased mass of platelets, leaving insufficient vWF for platelet adhesion. A mutation in the JAK2 kinase (V617F) is present in 40–50% of cases: it is diagnostic. JAK2 is a member of the Janus kinase family.
In 2013, two groups detected calreticulin mutations in a majority of JAK2-negative/MPL-negative patients with essential thrombocytosis and primary myelofibrosis, which makes CALR mutations the second most common in myeloproliferative neoplasms. All mutations (insertions or deletions) affected the last exon, generating a reading frame shift of the resulting protein, that creates a novel terminal peptide and causes a loss of endoplasmic reticulum KDEL retention signal.
Diagnostic criteria
The following revised diagnostic criteria for essential thrombocythaemia were proposed in 2005. The diagnosis requires the presence of both A criteria together with B3 to B6, or of criterion A1 together with B1 to B6. The criteria is as follows:
Indications
Not all those affected will require treatment at presentation. People are usually split up into low and high risk for bleeding/blood clotting groups (based on their age, their medical history, their blood counts and their lifestyles), low risk individuals are usually treated with aspirin, whereas those at high risk are given hydroxycarbamide and/or other treatments that reduce platelet count (such as interferon-α and anagrelide).
Agents
Hydroxycarbamide, interferon-α and anagrelide can lower the platelet count. Low-dose aspirin is used to reduce the risk of thrombosis unless the platelet count is very high, where there is a risk of bleeding from the disease and hence this measure would be counter-productive (as they increase one's risk for bleeds).
The PT1 study compared hydroxyurea plus aspirin to anagrelide plus aspirin as initial therapy for ET. Hydroxyurea treated patients had a lower incidence of arterial thrombosis, lower incidence of severe bleeding and lower incidence of transformation to myelofibrosis, but the risk of venous thrombosis was higĺ hydroxycarbamide than with anagrelide. It is unknown whether the results are applicable to all ET patients.
Prognosis
Essential thrombocytosis is sometimes described as a slowly progressive disorder with long asymptomatic periods punctuated by thrombotic or haemorrhagic events. However, well-documented medical regimens can reduce and control the number of platelets, which reduces the risk of these thrombotic or haemorrhagic events. The lifespan of a well controlled ET person is well within the expected range for a person of similar age but without ET.
Epidemiology
The incidence of ET is 0.6-2.5/100,000 per year, the median age at onset is 65–70 years and it is more frequent in females than in males. The incidence in children is 0.09/100,000 per year.
Pregnancy
Hydroxycarbamide and anagrelide are contraindicated during pregnancy and nursing. Essential thrombocytosis can be linked with a three-fold increase in risk of miscarriage. Throughout pregnancy, close monitoring of the mother and fetus is recommended. Low-dose low molecular weight heparin (e.g. enoxaparin) may be used. For life-threatening complications, the platelet count can be reduced rapidly using platelet apheresis, a procedure that removes platelets from the blood and returns the remainder to the patient.