
 | ||
In evolutionary biology and population genetics, the error threshold (or critical mutation rate) is a limit on the number of base pairs a self-replicating molecule may have before mutation will destroy the information in subsequent generations of the molecule. The error threshold is crucial to understanding "Eigen's paradox".
Contents
The error threshold is a concept in the origins of life (abiogenesis), in particular of very early life, before the advent of DNA. It is postulated that the first self-replicating molecules might have been small ribozyme-like RNA molecules. These molecules consist of strings of base pairs or "digits", and their order is a code that directs how the molecule interacts with its environment. All replication is subject to mutation error. During the replication process, each digit has a certain probability of being replaced by some other digit, which changes the way the molecule interacts with its environment, and may increase or decrease its fitness, or ability to reproduce, in that environment.
Fitness landscape
It was noted by Manfred Eigen in his 1971 paper (Eigen 1971) that this mutation process places a limit on the number of digits a molecule may have. If a molecule exceeds this critical size, the effect of the mutations become overwhelming and a runaway mutation process will destroy the information in subsequent generations of the molecule. The error threshold is also controlled by the fitness landscape for the molecules. Molecules that differ only by a few mutations may be thought of as "close" to each other, while those that differ by many mutations are distant from each other. Molecules that are very fit, and likely to reproduce, have a "high" fitness, those less fit have "low" fitness.
These ideas of proximity and height form the intuitive concept of the "fitness landscape". If a particular sequence and its neighbors have a high fitness, they will form a quasispecies and will be able to support longer sequence lengths than a fit sequence with few fit neighbors, or a less fit neighborhood of sequences. Also, it was noted by Wilke (Wilke 2005) that the error threshold concept does not apply in portions of the landscape where there are lethal mutations, in which the induced mutation yields zero fitness and prohibits the molecule from reproducing.
Eigen's Paradox
Eigen's paradox is one of the most intractable puzzles in the study of the origins of life. It is thought that the error threshold concept described above limits the size of self replicating molecules to perhaps a few hundred digits, yet almost all life on earth requires much longer molecules to encode their genetic information. This problem is handled in living cells by enzymes that repair mutations, allowing the encoding molecules to reach sizes on the order of millions of base pairs. These large molecules must, of course, encode the very enzymes that repair them, and herein lies Eigen's paradox, first put forth by Manfred Eigen in his 1971 paper (Eigen 1971). Simply stated, Eigen's paradox amounts to the following:
This is a chicken-or-egg kind of a paradox, with an even more difficult solution. Which came first, the large genome or the error correction enzymes? A number of solutions to this paradox have been proposed:
A simple mathematical model
Consider a 3-digit molecule [A,B,C] where A, B, and C can take on the values 0 and 1. There are eight such sequences ([000], [001], [010], [011], [100], [101], [110], and [111]). Let's say that the [000] molecule is the most fit; upon each replication it produces an average of
Note that the number of sequences for distance d is just the binomial coefficient
where the fitness matrix w according to quasispecies model is given by:
where
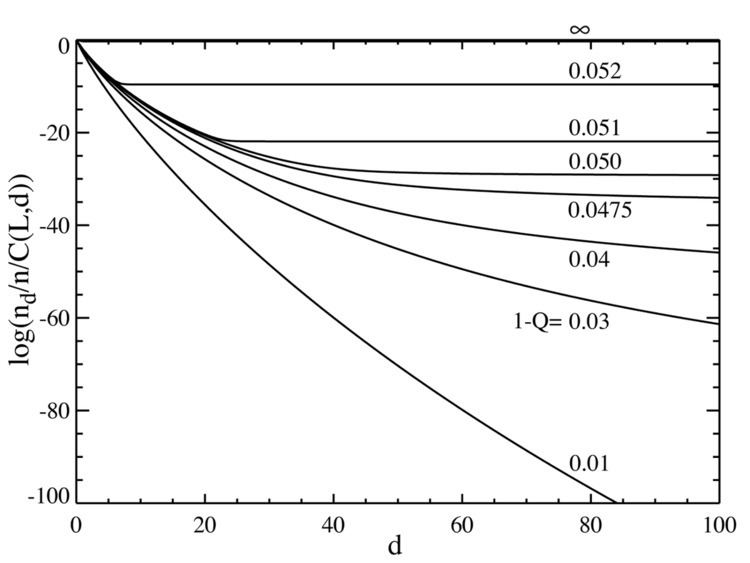
If we now go to the case where the number of base pairs is large, say L=100, we obtain behavior that resembles a phase transition. The plot below on the left shows a series of equilibrium concentrations divided by the binomial coefficient
It can be seen that there is a sharp transition at a value of 1-Q just a bit larger than 0.05. For mutation rates above this value, the population of the master sequence drops to practically zero. Above this value, it dominates.
In the limit as L approaches infinity, the system does in fact have a phase transition at a critical value of Q: