
ICD-9-CM 757.39 MedlinePlus 001457 | ICD-10 Q81 DiseasesDB 31928 33248 eMedicine derm/124 | |
 | ||
Epidermolysis bullosa (EB) is a group of inherited connective tissue diseases that cause blisters in the skin and mucosal membranes, with an incidence of 20 per million newborns in the United States. It is a result of a defect in anchoring between the epidermis and dermis, resulting in friction and skin fragility. Its severity ranges from mild to lethal.
Contents
- Classification
- Epidermolysis bullosa simplex
- Junctional epidermolysis bullosa
- Dystrophic epidermolysis bullosa
- Other
- Pathophysiology
- Treatment
- Prognosis
- Epidemiology
- Monitoring
- References
The condition was brought to public attention in 2004 in the UK through the Channel 4 documentary The Boy Whose Skin Fell Off, chronicling the life and death of Jonny Kennedy, an Englishman with EB. In the United States, the same could be said of the HBO documentary My Flesh and Blood from 2003.
"Butterfly Children" is a term often used to describe younger patients (because the skin is said to be as fragile as a butterfly’s wings), "Cotton Wool Babies", or (in South America) as "Crystal Skin Children".
Classification
Epidermolysis bullosa refers to a group of inherited disorders that involve the formation of blisters following trivial trauma. Over 300 mutations have been identified in this condition. They have been classified into the following types:
Epidermolysis bullosa simplex
Epidermolysis bullosa simplex is a form of epidermolysis bullosa that causes blisters at the site of rubbing. It typically affects the hands and feet, and is typically inherited in an autosomal dominant manner, affecting the keratin genes KRT5 and KRT14.
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa is an inherited disease affecting laminin and collagen. This disease is characterised by blister formation within the lamina lucida of the basement membrane zone and is inherited in an autosomal recessive manner. It also presents with blisters at the site of friction, especially on the hands and feet, and has variants that can occur in children and adults. Less than one person per million people is estimated to have this form of epidemolysis bullosa.
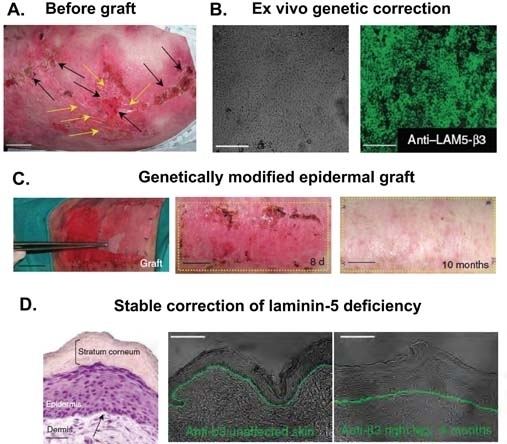
Dystrophic epidermolysis bullosa
Dystrophic epidermolysis bullosa is an inherited variant affecting the skin and other organs. "Butterfly children" is the term given to those born with the disease, as their skin is seen to be as delicate and fragile as a butterfly's wings. Dystrophic epidermolysis bullosa is caused by genetic defects (or mutations) within the human COL7A1 gene encoding the protein type VII collagen (collagen VII). DEB-causing mutations can be either autosomal dominant or autosomal recessive.
Other
Pathophysiology
The human skin consists of two layers: an outermost layer called the epidermis and a layer underneath called the dermis. In individuals with healthy skin, there are protein anchors between these two layers that prevent them from moving independently from one another (shearing). In people born with EB, the two skin layers lack the protein anchors that hold them together, resulting in extremely fragile skin—even minor mechanical friction (like rubbing or pressure) or trauma will separate the layers of the skin and form blisters and painful sores. Sufferers of EB have compared the sores with third-degree burns. Furthermore, as a complication of the chronic skin damage, people suffering from EB have an increased risk of malignancies (cancers) of the skin.
Treatment
Recent research has focused on changing the mixture of keratins produced in the skin. There are 54 known keratin genes—of which 28 belong to the type I intermediate filament genes and 26 to type II—which work as heterodimers. Many of these genes share substantial structural and functional similarity, but they are specialized to cell type and/or conditions under which they are normally produced. If the balance of production could be shifted away from the mutated, dysfunctional keratin gene toward an intact keratin gene, symptoms could be reduced. For example, sulforaphane, a compound found in broccoli, was found to reduce blistering in a mouse model to the point where affected pups could not be identified visually, when injected into pregnant mice (5 µmol/day = 0.9 mg) and applied topically to newborns (1 µmol/day = 0.2 mg in jojoba oil).
As of 2008 clinical research at the University of Minnesota has included a bone marrow transplant to a 2-year-old child who is one of 2 brothers with EB. The procedure was successful, strongly suggesting that a cure may have been found. A second transplant has also been performed on the child's older brother, and a third transplant is scheduled for a California baby. The clinical trial will ultimately include transplants to 30 subjects. However, the severe immunosuppression that bone marrow transplantation requires causes a significant risk of serious infections in patients with large scale blisters and skin erosions. Indeed, at least four patients have died in the course of either preparation for or institution of bone marrow transplantation for epidermolysis bullosa, out of only a small group of patients treated so far.
A pilot study performed in 2015 suggests that systemic granulocyte-colony stimulating factor (G-CSF) may promote increased wound healing in patients with dystrophic epidermolysis bullosa. In this study seven patients with dystrophic epidermolysis bullosa were treated daily with subcutaneous G-CSF for six days and then re-evaluated on the seventh day. After six days of treatment with G-CSF, the size of the open lesions were reduced by a median of 75.5% and the number of blisters and erosions on the patients were reduced by a median of 36.6%.
Prognosis
A 2014 study classified cases into three types -- epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), and dystrophic epidermolysis bullosa (DEB) -- and reviewed their times of death. The first two types tended to die in infancy and the last in early adulthood.
Epidemiology
An estimated 20 per million live births are diagnosed with EB, and 9 per million people in the general population have the condition. Of these cases, approximately 92% are epidermolysis bullosa simplex (EBS), 5% are dystrophic epidermolysis bullosa (DEB), 1% are junctional epidermolysis bullosa (JEB), and 2% are unclassified. Carrier frequency ranges from 1 in 333 for JEB, to 1 in 450 for DEB; the carrier frequency for EBS is presumed to be much higher than JEB or DEB.
The disorder occurs in every racial and ethnic group and affects both sexes.
Monitoring
The Epidermolysis Bullosa Activity and Scarring index (EBDASI) is a scoring system that objectively quantifies the severity of epidermolysis bullosa. The EBDASI is a tool for clinicians and patients to monitor the severity of the disease. It has also been designed to evaluate the response to new therapies for the treatment of EB. The EBDASI was developed and validated by Professor Dedee Murrell and her team of students and fellows at the St George Hospital, University of New South Wales, in Sydney, Australia. It was presented at the International Investigative Dermatology congress in Edinburgh in 2013 and a paper-based version was published in the Journal of American Academy of Dermatology in 2014.