
CAS Number 140111-52-0 DrugBank DB07720 KEGG C11690 Formula C11H13ClN2 | PubChem CID 11031065 ChemSpider 10399316 ChEMBL CHEMBL298826 Molar mass 208.69 g/mol | |
 | ||
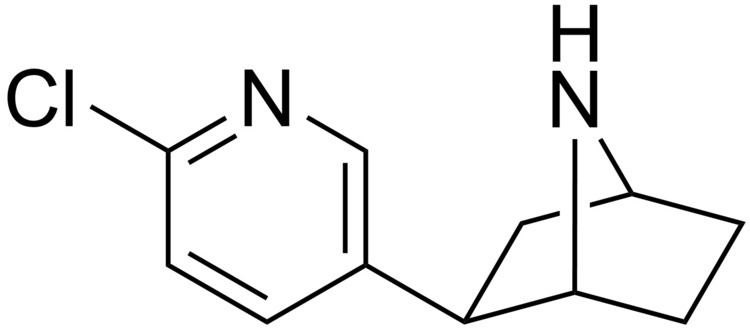
Epibatidine is an alkaloid that is secreted by the Ecuadoran frog Epipedobates anthonyi. It was discovered by John W. Daly in 1974, but its structure was not fully elucidated until 1992.
Contents
- History
- Synthesis
- Synthetic analogs
- Chemical structure
- Mechanism of action
- Symptoms
- Pharmacology
- Potential medical uses
- Antidote
- References
Epibatidine is toxic. Its toxicity stems from its ability to interact with nicotinic and muscarinic acetylcholine receptors. These receptors are involved in the transmission of painful sensations, and in movement, among other functions. Epibatidine then causes numbness and eventually paralysis. Doses are lethal when the paralysis causes respiratory arrest. Originally, it was thought that epibatidine could be useful as a drug. However, because it can be deadly even at very low doses, it is no longer being researched for potential therapeutic uses.
History
Epibatidine was discovered by John W. Daly in 1974. It was isolated from the skin of Epipdobates anthonyi frogs collected by Daly and colleague, Charles Myers. Between 1974 and 1979, Daly and Myers collected the skins of nearly 3000 frogs from various sites in Ecuador, after finding that a small injection of a preparation from their skin caused analgesic (painkilling) symptoms in mice with symptoms that resembled those of an opioid. Despite its common name - Anthony’s Poison Arrow frog - suggesting that it was used by natives when hunting, a paper written by Daly in 2000 claimed that there was no local folklore or folk medicine surrounding the frogs and that they were considered largely unimportant by the locals.

The structure of epibatidine was elucidated in 1992, an effort hindered by E. anthonyi gaining IUCN protected status in 1984. Furthermore, these frogs do not produce the toxin when bred and reared in captivity, because they do not synthesize epibatidine themselves. Like other poison dart frogs, they instead obtain it through their diet and then sequester it on their skin. Likely dietary sources are beetles, ants, mites, and flies. Overcoming the difficulties, the structure was eventually determined, and the first synthesis of epibatidine was completed in 1993. Many other synthesis methods have been developed since.
Because of its analgesic effect, there was intense interest in epibatidine’s use as a drug, because it was found not to be an opioid. This meant that it could potentially be used without fear of addiction. However, it was soon found that it cannot be used in humans because the dose resulting in toxic symptoms is too low for it to be safe.
Synthesis
Several total synthesis routes have been devised due to the relative scarcity of epibatidine in nature.
After the discovery of the structure of epibatidine, more than fifty ways to synthesize it in the laboratory have been devised. In the first reported example, a nine-step procedure produces the substance as a racemate (in contrast, the naturally occurring compound is the (+)-enantiomer; the (−)-enantiomer does not occur naturally). It was later determined that the (+) and (-) enantiomers had equivalent analgesic as well as toxic effects. The process has proven to be quite productive, with a yield of about 40%.
An enantioselective synthesis reported by E J Corey starting from chloronicotinaldehyde is outlined below:
In addition to Corey's method, other notable methods include those of Broka, Huang and Shen, and Clayton and Regan.
Synthetic analogs
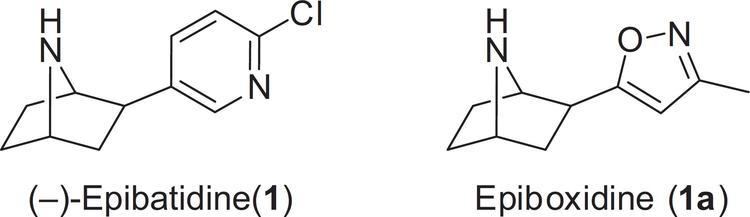
A number of approaches to discovering structural analogs of epibatine that maintain analgesics effects, but without the toxicity, have been attempted. For example, Abbott Laboratories has produced derivatives of epibatidine including ABT-594. Other epibatidine include ABT-418 and epiboxidine.
No attempt to prepare a derivative of epibatidine with reduced toxicity and an effective analgesic effect has yet been successful.
Chemical structure
Epibatidine is a pyridine with a structure similar to that of nicotine. It is a hygroscopic oily substance which is a base.
Mechanism of action
Epibatidine has two mechanisms of action: it can bind to either nicotinic acetylcholine receptors (nAChR) or muscarinic acetylcholine receptors (mAChR). Specifically, the analgesic property of epibatidine is believed to take place by its binding to the α4/β2 subtype of nicotinic receptors. Epibatidine also binds to the α3/β4 subtype and to a much lesser extent α7 receptors (affinity 300-fold less than for α4/β2) The rank order of affinities is αε > αγ > αδ.
Nicotinic acetylcholine receptors are found in the post-synaptic membranes of nerve cells. They propagate neurotransmission in the central and peripheral nervous system. When neurotransmitters bind to these receptors, ion channels open, allowing Na+ and Ca2+ ions to move across the membrane. This depolarizes the post-synaptic membrane, inducing an action potential that propagates the signal. This signal will ultimately induce release of dopamine and norepinephrine, resulting in an antinociceptive effect on the organism. The usual neurotransmitter for nAChR is acetylcholine. However, other substances (such as epibatidine and nicotine) are also able to bind to the receptor and induce a similar, if not identical, response. Epibatidine has an extremely high affinity for nAChRs and will induce a response at concentrations of ~10 µM. This is a 1000 times lower concentration when compared to a nicotine-induced response.
The paralytic property of epibatidine takes place after its binding to muscarinic acetylcholine receptors (mAChR). These receptors are G-protein coupled of five subtypes. The M2 and M4 subytpes are coupled to an inhibitory protein which impedes the functioning of adenylyl cyclase. This enzyme catalyzes the transformation of ATP into cAMP, which is an important cellular second messenger.
The M1, M3 and M5 subytpes are coupled to a Gq-protein, which activate phosphatidylinositol 3-kinases (PI3K). These enzymes, when activated, catalyze the reaction of Phosphatidylinositol 4,5-bisphosphate (PIP2)into diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3). These second messengers can affect several processes in the cell, such as (sarco) endoplasmic reticulum calcium ATP-ase (SERCA).
Low doses of epibatidine will only affect the nAChRs, due to a higher affinity to nAChRs than to mAChRs. Higher doses, however, will cause epibatidine to bind to the mAChRs and result in paralytic effects in humans.
Both (+)- and (-)-enantiomers of epibatidine are biologically active, and both have similar binding affinities to nAChRs Only the (+)-enantiomer does not induce tolerance, its chief advantage over morphine.
Symptoms
Epibatidine has several toxic consequences. Empirically proven effects include splanchnic sympathetic nerve discharge and increased arterial pressure. The nerve discharge effects can cause antinociception partially mediated by agonism of central nicotinic acetylcholine receptors at low doses of epibatidine; 5 µg/kg. At higher doses, however, epibatidine will cause paralysis and loss of consciousness, coma and eventually death. The median lethal dose (LD50) of epibatidine lies between 1.46 µg/kg and 13.98 µg/kg. This makes epibatidine somewhat more toxic than dioxin (with an average LD50 of 22.8 µg/kg). Due to the small difference between its toxic concentration and antinociceptive concentration, its therapeutic uses are very limited.
In research on mice, administration of doses greater than 5 μg/kg of epibatidine caused a dose-dependent paralyzing effect on the organism. With doses over 5 μg/kg, symptoms included hypertension (increased blood pressure), paralysis in the respiratory system, seizures, and, ultimately, death. The symptoms do, however, change drastically when lower doses are given. Mice became resistant to pain and heat with none of the negative effects of higher doses.
Pharmacology
Epibatidine most effectively enters the body through injection. Oddly enough, in vitro studies seem to suggest that epibatidine is hardly, if at all, metabolized in the human body.
Also there is currently little information on the path of clearance from the body. Maximum concentration in the brain is reached at about 30 minutes after injection and epibatidine is still present after four hours, showing that clearance is slow.
Potential medical uses
Epibatidine has a high analgesic potency, as stated above. Studies show it has a potency at least 200 times that of morphine. As the compound was not addictive nor did it cause habituation,, it was initially thought to be very promising to replace morphine as a painkiller. However, the therapeutic concentration is very close to the toxic concentration. This means that even at a therapeutic dose (5 µg/kg), some epibatidine might bind to the muscarinic acetylcholine receptors and cause adverse effects, such as hypertension, bradycardia and muscular paresis.
Compared to the gold standard in pain management, morphine, epibatidine needed only 2.5 μg/kg to initiate a pain-relieving effect whilst the same effect required approximately 10 mg/kg of morphine (4,000 times the efficacy.) Currently, only rudimentary research into epibatidine's effects has yet been performed; the drug has been administered only to rodents for analysis at this time.
Antidote
The antidote to epibatidine is mecamylamine, a nicotinic acetylcholine receptor antagonist that is non-selective and non-competitive.