
 | ||
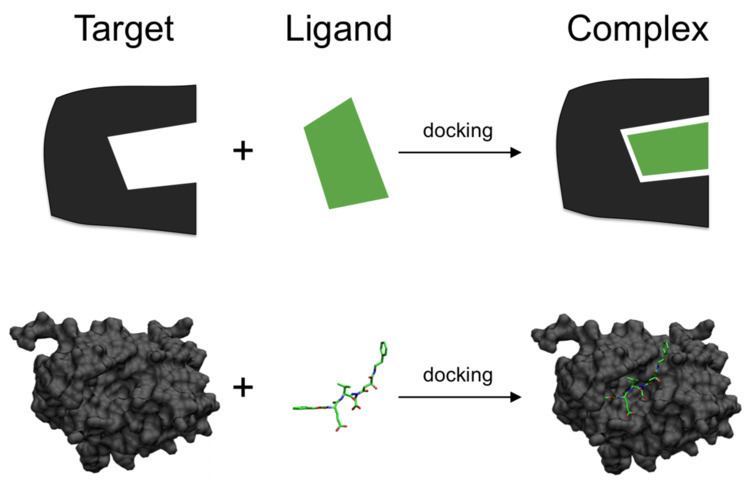
In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
Contents
- Definition of problem
- Docking approaches
- Shape complementarity
- Simulation
- Mechanics of docking
- Search algorithm
- Ligand flexibility
- Receptor flexibility
- Scoring function
- Docking assessment
- Docking accuracy
- Enrichment factor
- Prospective
- Benchmarking
- Applications
- List of protein ligand docking software
- References
The associations between biologically relevant molecules such as proteins, nucleic acids, carbohydrates, and lipids play a central role in signal transduction. Furthermore, the relative orientation of the two interacting partners may affect the type of signal produced (e.g., agonism vs antagonism). Therefore, docking is useful for predicting both the strength and type of signal produced.
Molecular docking is one of the most frequently used methods in structure-based drug design, due to its ability to predict the binding-conformation of small molecule ligands to the appropriate target binding site. Characterisation of the binding behaviour plays an important role in rational design of drugs as well as to elucidate fundamental biochemical processes.
Definition of problem
One can think of molecular docking as a problem of “lock-and-key”, in which one wants to find the correct relative orientation of the “key” which will open up the “lock” (where on the surface of the lock is the key hole, which direction to turn the key after it is inserted, etc.). Here, the protein can be thought of as the “lock” and the ligand can be thought of as a “key”. Molecular docking may be defined as an optimization problem, which would describe the “best-fit” orientation of a ligand that binds to a particular protein of interest. However, since both the ligand and the protein are flexible, a “hand-in-glove” analogy is more appropriate than “lock-and-key”. During the course of the docking process, the ligand and the protein adjust their conformation to achieve an overall "best-fit" and this kind of conformational adjustment resulting in the overall binding is referred to as "induced-fit".
Molecular docking research focusses on computationally simulating the molecular recognition process. It aims to achieve an optimized conformation for both the protein and ligand and relative orientation between protein and ligand such that the free energy of the overall system is minimized.
Docking approaches
Two approaches are particularly popular within the molecular docking community. One approach uses a matching technique that describes the protein and the ligand as complementary surfaces. The second approach simulates the actual docking process in which the ligand-protein pairwise interaction energies are calculated. Both approaches have significant advantages as well as some limitations. These are outlined below.
Shape complementarity
Geometric matching/ shape complementarity methods describe the protein and ligand as a set of features that make them dockable. These features may include molecular surface / complementary surface descriptors. In this case, the receptor’s molecular surface is described in terms of its solvent-accessible surface area and the ligand’s molecular surface is described in terms of its matching surface description. The complementarity between the two surfaces amounts to the shape matching description that may help finding the complementary pose of docking the target and the ligand molecules. Another approach is to describe the hydrophobic features of the protein using turns in the main-chain atoms. Yet another approach is to use a Fourier shape descriptor technique. Whereas the shape complementarity based approaches are typically fast and robust, they cannot usually model the movements or dynamic changes in the ligand/ protein conformations accurately, although recent developments allow these methods to investigate ligand flexibility. Shape complementarity methods can quickly scan through several thousand ligands in a matter of seconds and actually figure out whether they can bind at the protein’s active site, and are usually scalable to even protein-protein interactions. They are also much more amenable to pharmacophore based approaches, since they use geometric descriptions of the ligands to find optimal binding.
Simulation
Simulating the docking process as such is much more complicated. In this approach, the protein and the ligand are separated by some physical distance, and the ligand finds its position into the protein’s active site after a certain number of “moves” in its conformational space. The moves incorporate rigid body transformations such as translations and rotations, as well as internal changes to the ligand’s structure including torsion angle rotations. Each of these moves in the conformation space of the ligand induces a total energetic cost of the system. Hence, the system's total energy is calculated after every move.
The obvious advantage of docking simulation is that ligand flexibility is easily incorporated, whereas shape complementarity techniques must use ingenious methods to incorporate flexibility in ligands. Also, it more accurately models reality, whereas shape complimentary techniques are more of an abstraction.
Clearly, simulation is computationally expensive, having to explore a large energy landscape. Grid-based techniques, optimization methods, and increased computer speed have made docking simulation more realistic.
Mechanics of docking
To perform a docking screen, the first requirement is a structure of the protein of interest. Usually the structure has been determined using a biophysical technique such as x-ray crystallography or NMR spectroscopy, but can also derive from homology modeling construction. This protein structure and a database of potential ligands serve as inputs to a docking program. The success of a docking program depends on two components: the search algorithm and the scoring function.
Search algorithm
The search space in theory consists of all possible orientations and conformations of the protein paired with the ligand. However, in practice with current computational resources, it is impossible to exhaustively explore the search space—this would involve enumerating all possible distortions of each molecule (molecules are dynamic and exist in an ensemble of conformational states) and all possible rotational and translational orientations of the ligand relative to the protein at a given level of granularity. Most docking programs in use account for the whole conformational space of the ligand (flexible ligand), and several attempt to model a flexible protein receptor. Each "snapshot" of the pair is referred to as a pose.
A variety of conformational search strategies have been applied to the ligand and to the receptor. These include:
Ligand flexibility
Conformations of the ligand may be generated in the absence of the receptor and subsequently docked or conformations may be generated on-the-fly in the presence of the receptor binding cavity, or with full rotational flexibility of every dihedral angle using fragment based docking. Force field energy evaluation are most often used to select energetically reasonable conformations, but knowledge-based methods have also been used.
Receptor flexibility
Computational capacity has increased dramatically over the last decade making possible the use of more sophisticated and computationally intensive methods in computer-assisted drug design. However, dealing with receptor flexibility in docking methodologies is still a thorny issue. The main reason behind this difficulty is the large number of degrees of freedom that have to be considered in this kind of calculations. Neglecting it, however, leads to poor docking results in terms of binding pose prediction.
Multiple static structures experimentally determined for the same protein in different conformations are often used to emulate receptor flexibility. Alternatively rotamer libraries of amino acid side chains that surround the binding cavity may be searched to generate alternate but energetically reasonable protein conformations.
Scoring function
Docking programs generate a large number of potential ligand poses, of which some can be immediately rejected due to clashes with the protein. The remainder are evaluated using some scoring function, which takes a pose as input and returns a number indicating the likelihood that the pose represents a favorable binding interaction and ranks one ligand relative to another.
Most scoring functions are physics-based molecular mechanics force fields that estimate the energy of the pose within the binding site. The various contributions to binding can be written as an additive equation:
The components consist of solvent effects, conformational changes in the protein and ligand, free energy due to protein-ligand interactions, internal rotations, association energy of ligand and receptor to form a single complex and free energy due to changes in vibrational modes. A low (negative) energy indicates a stable system and thus a likely binding interaction.
An alternative approach is to derive a knowledge-based statistical potential for interactions from a large database of protein-ligand complexes, such as the Protein Data Bank, and evaluate the fit of the pose according to this inferred potential.
There are a large number of structures from X-ray crystallography for complexes between proteins and high affinity ligands, but comparatively fewer for low affinity ligands as the later complexes tend to be less stable and therefore more difficult to crystallize. Scoring functions trained with this data can dock high affinity ligands correctly, but they will also give plausible docked conformations for ligands that do not bind. This gives a large number of false positive hits, i.e., ligands predicted to bind to the protein that actually don't when placed together in a test tube.
One way to reduce the number of false positives is to recalculate the energy of the top scoring poses using (potentially) more accurate but computationally more intensive techniques such as Generalized Born or Poisson-Boltzmann methods.
Docking assessment
The interdependence between sampling and scoring function affects the docking capability in predicting plausible poses or binding affinities for novel compounds. Thus, an assessment of a docking protocol is generally required (when experimental data is available) to determine its predictive capability. Docking assessment can be performed using different strategies, such as:
Docking accuracy
Docking accuracy represents one measure to quantify the fitness of a docking program by rationalizing the ability to predict the right pose of a ligand with respect to that experimentally observed.
Enrichment factor
Docking screens can be also evaluated by the enrichment of annotated ligands of known binders from among a large database of presumed non-binding, “decoy” molecules. In this way, the success of a docking screen is evaluated by its capacity to enrich the small number of known active compounds in the top ranks of a screen from among a much greater number of decoy molecules in the database. The area under the receiver operating characteristic (ROC) curve is widely used to evaluate its performance.
Prospective
Resulting hits from docking screens are subjected to pharmacological validation (e.g. IC50, affinity or potency measurements). Only prospective studies constitute conclusive proof of the suitability of a technique for a particular target.
Benchmarking
The potential of docking programs to reproduce binding modes as determined by X-ray crystallography can be assed by a range of docking benchmark sets.
For small molecules, several benchmark data sets for docking and virtual screening exist e.g. Astex Diverse Set consisting of high quality protein−ligand X-ray crystal structures or the Directory of Useful Decoys (DUD) for evaluation of virtual screening performance.
An evaluation of docking programs for their potential to reproduce peptide binding modes can be assessed by Lessons for Efficiency Assessment of Docking and Scoring (LEADS-PEP).
Applications
A binding interaction between a small molecule ligand and an enzyme protein may result in activation or inhibition of the enzyme. If the protein is a receptor, ligand binding may result in agonism or antagonism. Docking is most commonly used in the field of drug design — most drugs are small organic molecules, and docking may be applied to:
List of protein-ligand docking software
The number of docking programs currently available is high and has been steadily increasing over the last decades. The following list presents an overview of the most common protein-ligand docking programs, listed alphabetically, with indication of the corresponding year of publication, involved organisation or institution, short description, availability of a webservice and the license. This table is comprehensive but not complete.