
 | ||
Decarboxylative cross coupling reactions are chemical reactions in which a stable, substituted carboxylic acid is reacted with a halide (R–X, R=aryl, alkyl, etc.) in the presence of a metal catalyst, base and oxidant to form a new carbon-carbon bond (with loss of carbon dioxide).
Contents
- Copper monometallic systems
- Palladium monometallic systems
- Palladium copper bimetallic systems
- Palladiumsilver bimetallic systems
- Product scope via variation of substrates
- Biaryl formation
- Aryl alkynes
- Aryl ketones
- Aryl esters
- sp3C carboxylic acids
- Dienes and trienes
- Olefins via Heck type
- Phenanthrene derivatives
- CN coupling
- C S Coupling
- CP coupling
- CX coupling
- Decarboxylative Heck type
- Biaryl synthesis via Redox Neutral decarboxylative cross coupling
- Heteroaromatic acid coupling
- References
A significant advantage of this reaction is that it uses relatively inexpensive carboxylic acids (or their salts) and is far less air and moisture sensitive in comparison to typical cross-coupling organometallic reagents. Furthermore, the carboxylic acid moiety is a common feature of natural products and can also be prepared by relatively benign air oxidations. Additional benefits include the broad tolerance of functional groups, as well as the capacity to avoid the use of strong bases. An important elementary step in this reaction is protodecarboxylation or metallation to first convert the C–COOH bond to a C–H or C–M bond respectively.
Copper monometallic systems
The first reported decarboxylative cross coupling reaction was an Ullmann reaction, in 1966 by Nilsson et al. Thermal decarboxylation of copper benzoates, in the presence of an aryl halide, was found to produce (both symmetric and unsymmetric) biaryls through aryl-Cu intermediates.
This monometallic copper system required drastic conditions for complete cross-coupling, and had various intrinsic limitations, both of which prevented development of a catalytic, preparatory version of this reaction. It was not until 2009 that Liu and Shang et al. found that decarboxylative cross-coupling of aryl bromides and iodides with potassium polyfluorobenzoates could be achieved using monometallic copper iodide as a catalyst. The oxidative addition step was determined to be the rate-limiting step in the copper-only catalyst cycle (a contrast with Pd-catalyzed decarboxylative cross-coupling).
Cu(I)-only systems have also been found to promote coupling of alkynyl carboxylic acids with aryl halides (see aryl alkynes below), as well as decarboxylative dehydrogenative cross-coupling of amino acids with alkynes (or similar nucleophiles).
Catalysts for decarboxylative cross-coupling are of the general form ML2, with a wide variety of ligand types optimized for different substrates. Copper (and silver) centers are often complexed with phenanthrolines, and activity is reported to increase with electron-rich substituents on the ligands.
Palladium monometallic systems
In 2000, Steglich et al. reported an intramolecular Pd(II)-mediated decarboxylative cross-coupling reaction in their synthesis of lamellarin L. Myers et al. reported decarboxylative olefination of ortho-substituted arene carboxylates in the presence of an oxidant (Ag2CO3) in 2002.
Subsequent studies showed that homogenous Pd catalysts were able to decarboxylate acids at lower temperatures than their Cu and Ag counterparts, but were limited to electron rich ortho-substituted aromatic carboxylic acids. Despite this, palladium catalysts are able to promote a wide variety of cross-coupling reactions including biaryl formation and aryl alkyne formation, along with a variety of cross-coupling reactions in which the carboxylic acid is not bonded to an aromatic. Other Pd-catalyzed decarboxylation cross-coupling reactions include conjugated diene preparation (see dienes and trienes below) and dehydrogenative reactions (with a variety of substrate and catalyst combinations).
Contrarily to Cu-only systems, decarboxylative palladation is the rate-limiting step in the palladium catalytic cycle.
Palladium-–copper bimetallic systems
A Pd–Cu bimetallic system was not discovered until 2006 when Goossen et al. reported a decarboxylative cross-coupling of aryl halides with ortho-substituted aromatic carboxylic acids. Through subsequent studies it was found that the use of aryl triflates allowed substrate scope for cross-coupling to be extended to some aromatic carboxylates lacking any ortho-substitution (less reactive). This was a result of the fact that any halide anion generated in the reaction inhibited the Cu-catalyzed decarboxylation process. Further optimization of the system and catalyst conditions has made decarboxylative cross-coupling using bimetallic Pd–Cu systems applicable to organic synthesis, most predominantly in the formation of biaryls. As well, the variability of this combined catalytic system allows for promotion of a large spectrum of reactions, including aryl ketone formation, c-heteroatom cross-coupling, and many others.
Palladium–silver bimetallic systems
Silver being in the same group as copper, Pd–Ag(I) bimetallic systems are inherently similar to Pd–Cu catalytic systems. However, silver salts are better suited for protodecarboxylation of carboxylic acids than their copper equivalents, allowing milder reaction conditions in Pd–Ag cycles relative to Pd–Cu cycles. Ag(I) catalyzed monometallic systems have also been reported. Their proficiency (relative to copper) is likely attributed to lower electronegativity and greater expansion of d-orbitals, which promote decarboxylation of the substrate. One limitation of this catalyst combination is that the silver salts will form insoluble silver halides, forcing the reaction to require a stoichiometric amount of Ag if halides are present. This obstacle was overcome by Goossen et al. in 2010 by using aryl triflates, and catalytic reaction with aryl sulfonates has also been reported.
Product scope via variation of substrates
The product scope of this reaction is extremely broad with the use of different substrates; however development of different functionalities has required accompanied studies to determine the proper catalyst system. The most typical class of reactions involves coupling between C–COOH and C–X bonds, however C–COOH and C–M cross-coupling, homo-coupling of carboxylic acids, heck coupling, and dehydrogenative cross-coupling can also be including in this class as they release CO2. Heteroatom cross coupling reactions involving formation of C–N, C–S, C–P, and C–X bonds have also been demonstrated.
Biaryl formation
Per IUPAC, the term biaryl refers to an assembly of two aromatic rings joined by a single bond, starting with the simplest, biphenyl. Biaryls constitute an important structural motif of physical organic, synthetic, and catalytic interest—for instance, underlying the area of atropisomers in enantioselective synthesis—and they appear in many pharmaceutical, agrochemical, and materials (e.g. LCD) applications. The example of a coupling reaction reaction used in their preparation is an alternative to the traditional Suzuki and Stille cross-coupling reactions, and various catalysts have been employed for this transformation; Goossen et al. reported the formation of biaryls from palladium and copper-catalzyed cross-coupling reactions of an aryl or heteroaryl carboxylic acid and an aryl halide (I, Br, or Cl) in the presence of a base.
Aryl alkynes
Aryl alkynes are typically made utilizing the Sonogashira reaction which is the palladium catalyzed cross-coupling reaction of terminal alkynes and aryl halides. Instead of the terminal alkynes, alkyne carboxylic acids has advantages, easy handling and storage. The first decarboxylative coupling of alkyne carboxylic acids was reported in 2008 by S. Lee. They employed propiolic acid as an alkyne source. One year later, S. Lee applied the decarboxylative coupling reactions toward 2-octynoic acid and phenylpropiolic acid. In 2010, Xue et al. reported the coupling of an aryl halide and alkynyl carboxylic acid under mild reactions conditions and a copper-only catalyst to obtain aryl alkynes.
Aryl ketones
Further work by Goossen et al. described the synthesis of ketones from α-oxocarboxylic acids with aryl or heteroaryl bromides through an acyl anion intermediate.
Aryl esters
Shang et al. discovered the decarboxylative coupling of potassium oxalate monoesters with aryl halides to obtain aryl or alkenyl esters.
sp3C carboxylic acids
Many decarboxylative cross coupling reactions involve the breaking of sp2C–COOH and spC–COOH bonds, therefore subsequent studies have attempted to enable cross coupling with sp3C carboxylic acids. One such reaction by Shang et al. described a palladium catalyzed cross coupling that enables the formation of functionalized pyridines, pyrazines, quinolines, benzothiazoles, and benzoxazoles. The position of the nitrogen atom in the '2' position relative to the linkage is found to be required, therefore implying its binding to Pd in a transition state.
Dienes and trienes
Miura et al. reported the cross coupling of vinyl bromides with an alkenyl carboxylic acid using a palladium catalyst. Some of the conjugated dienes prepared were reported to exhibit solid state fluorescence.
Olefins via Heck-type
A decarboxylative Heck coupling by Su et al. can be used to obtain an aryl olefin using benzoquinone as the oxidant.
Phenanthrene derivatives
Wang et al. proposed a novel method for [4+2] annulation via a palladium catalyzed intermolecular pathway. Derivatives are formed in moderate to good yield; acridine is essential for high reaction efficiency.
C–N coupling
Jiao et al. enabled the formation of a C–N bond via cross-coupling using air as an oxidant and a copper catalyst. No conditions are known for a C–N cross-coupling that breaks a sp3 or sp2 C–COOH bond.
C-S Coupling
Liu et al. reported the C-S coupling of aryl carboxylic acids with disulphides or thiols using a Pd/Cu catalyst system.
C–P coupling
Using either Pd–Cu or Cu cataylsts Yang et al. reported the first example of decarboxylative C–P cross-coupling.
C–X coupling
Wu et al. reported a C–X cross coupling using CuX2 (X= Br, Cl) and a silver catalyst to obtain aryl halides.
Decarboxylative Heck type
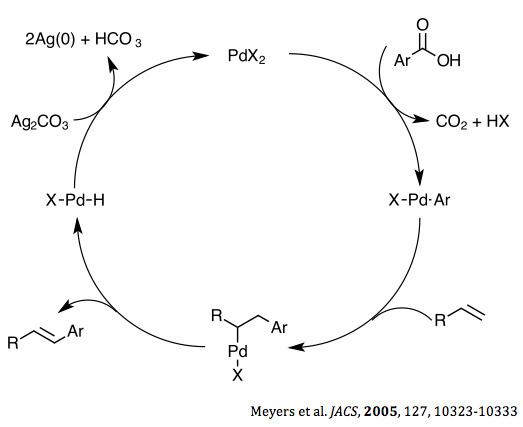
In 2005, Meyers et al. Proposed the following mechanism for the decarboxylative cross-coupling reaction. The initial and rate determining step is the decarboxylation. The ipso carbon of the arene ring is thought to coordinate to the palladium centre initially and is followed by the expulsion of carbon dioxide, forming an aryl–palladium intermediate. The olefin then inserts between the arene and palladium center, which then undergoes beta elimination to form the desired vinyl halide, as well as a palladium hydride. This proton is abstracted by silver carbonate, which acts as both a base and an oxidant to regenerate the starting palladium complex completing the catalytic cycle.
Biaryl synthesis via Redox-Neutral decarboxylative cross-coupling
In 2006 Goossen et al. proposed a reaction to synthesize biaryl compounds via catalytic decarboxylative cross coupling. The mechanism involves two overlapping cycles, one using a copper halide and the other using palladium. The decarboxylation step occurs between the substituted benzoic acid and copper halide to form the intermediate aryl copper species. The palladium initially undergoes oxidative addition from the aryl halide to form a Pd(II) aryl complex. After both of these initial steps, the substituted aryl copper undergoes trans-metallation with the palladium complex. This step forms the copper halide, which then undergoes anion exchange with the substituted benzoic acid to reform the aryl copper intermediate, continuing the catalytic cycle. The other complex formed in the trans-metallation step is a bis-aryl palladium(II), which then undergoes reductive elimination to form the desired bis-aryl species as well as the starting Pd(0) complex, thus completing the catalytic cycle.
Heteroaromatic acid coupling
Forgione, P., Bilodeau, F et al. reported that heteroatoms containing a carboxylic acid also are tolerated by palladium monometallic systems and undergo decarboxylative cross coupling with aryl halides. In the proposed mechanism the initial step is oxidative addition of the aryl halide forming an aryl–palladium intermediate. Electrophilic palladation then occurs at carbon-3 of the heteroatom. From this intermediate there are two possible pathways for the cycle to continue on. The first is palladium migration from carbon-3 to carbon-2 along with the expulsion of carbon dioxide. This forms the aryl–palladium–heteroatom intermediate, which undergoes reductive elimination to form the final heteroaromatic compound. The second pathway only occurs when R is a proton. If this is the case, deprotonation occurs to regain aromaticity of the heteroatom. This intermediate then undergoes reductive elimination, coupling the aryl to the carbon-3 position of the heteroatom. As this compound still contains the carboxylic acid it is then free to re enter the catalytic cycle where it undergoes coupling at the carbon 2 position, along with the expulsion of carbon dioxide to form a biaryl heteroatom. As this pathway competes with the decarboxylation step, two products are formed making this reaction less selective. As a result, heteroatoms, which are substituted at the carbon 3 position and are more favored due to the higher level of control they provide.