
 | ||
Crystal structure prediction and its application in earth and materials sciences
Crystal structure prediction (CSP) is the calculation of the crystal structures of solids from first principles. Reliable methods of predicting the crystal structure of a compound, based only on its molecular structure, has been a goal of the physical sciences since the 1950s. Computational methods employed include simulated annealing, evolutionary algorithms, distributed multipole analysis, random sampling, basin hopping, data mining, density functional theory and molecular mechanics.
Contents
Evolutionary methods for crystal structure prediction and alternatives
History
The crystal structures of simple ionic solids have long been rationalised in terms of Pauling's rules, first set out in 1929 by Linus Pauling. For metals and semiconductors one has different rules involving valence electron concentration. However, prediction and rationalization are rather different things. Most commonly, by crystal structure prediction one understands search for minimum-energy arrangement of the atoms (or, for molecular crystals, of the molecules) in space. The problem has two facets - combinatorial (the "search" problem, in practice most acute for inorganic crystals), and energetic ("ranking", most acute for molecular organic crystals). For complex non-molecular crystals ("search problem"), major recent advances have been the Martonak version of metadynamics, the Oganov-Glass evolutionary algorithm USPEX,. The latter is capable of solving the global optimization problem with up to a few hundred degrees of freedom, while the approach of metadynamics is to reduce all structural variables to a handful of "slow" collective variables (which often works).
Molecular crystals
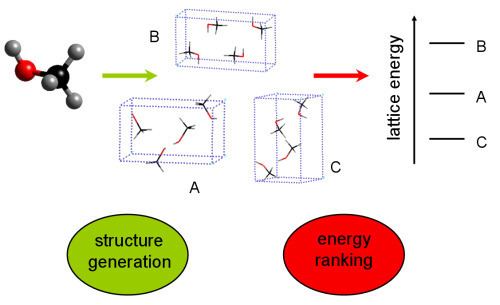
Predicting organic crystal structures is important to academic and industrial science, particularly for pharmaceuticals and pigments, where understanding polymorphism is beneficial. The crystal structures of molecular substances, particularly organic compounds, are very hard to predict and rank in stability. Intermolecular interactions are relatively weak and non-directional and long range. This results in typical lattice and free energy differences between polymorphs that are often only a few kJ/mol, very rarely exceeding 10 kJ/mol. Crystal structure prediction methods often locate many possible structures within this small energy range. These small energy differences are challenging to predict reliably and with a reasonable computational effort.
Since 2007, significant progress has been made in the CSP of small organic molecules, with several different methods proving effective. The most widely discussed method first ranks the energies of all possible crystal structures using a customised MM force field, and finishes by using a dispersion-corrected DFT step to estimate the lattice energy and stability of each short-listed candidate structure.
Crystal structure prediction software
The following codes can predict stable and metastable structures given chemical composition and external conditions (pressure, temperature):