
 | ||
Craniofrontonasal dysplasia (craniofrontonasal syndrome, craniofrontonasal dysostosis, CFND) is a very rare X-linked malformation syndrome caused by mutations in the ephrin-B1 gene (EFNB1). Phenotypic expression varies greatly amongst affected individuals, where females are more commonly and generally more severely affected than males.
Contents
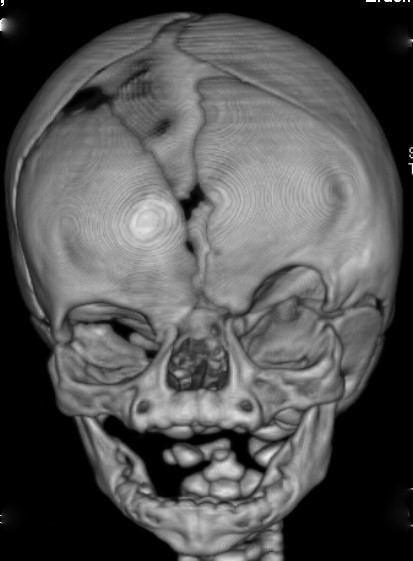
Common physical malformations are: craniosynostosis of the coronal suture(s), orbital hypertelorism, bifid nasal tip, dry frizzy curled hair, longitudinal ridging and/or splitting of the nails, and facial asymmetry.
The diagnosis CFND is determined by the presence of a mutation in the EFNB1 gene. Physical characteristics may play a supportive role in establishing the diagnosis.
The treatment is always surgical and is based on each patients specific phenotypic presentation.
Epidemiology
Craniofrontonasal dysplasia is a very rare genetic condition. As such there is little information and no consensus in the published literature regarding the epidemiological statistics.
The incidence values that were reported ranged from 1:100,000 to 1:120,000.
Phenotype
Phenotypic expression varies greatly between individuals with CFND. Some of the more prominent characteristics are:
Other characteristics that are less frequently seen are: broad nasal base, low anterior hair line, low set ears, crowding of the teeth, maxillary hypoplasia, rounded and sloping shoulders, pectus excavatum, scoliosis, high arched palate, orbital dystopia, low implant of the breasts with asymmetric nipples and volume, webbed neck, hand or foot abnormalities such as clinodactyly (most common is a curved 5th finger) and cutaneous syndactyly (webbed fingers / toes).
Females are more commonly and usually more severely affected than males. Males can however have (some of) the same symptoms as females, but this is not frequently seen. Most males have mild symptoms such as hypertelorism and a broad nasal base with bifid nose, but can also be a carrier of the mutation yet stay clinically unaffected.
Genetics and heritability
CFND is a very rare X-linked malformation syndrome caused by mutations in the ephrin-B1 gene (EFNB1). The EFNB1 gene codes for a membrane-anchored ligand which can bind to an ephrin tyrosine-kinase receptor. This ephrin receptor is, amongst other things, responsible for the regulation of embryonic tissue-border formation, and is important for skeletal and craniofacial development. As the ephrin receptor and its EFNB1 ligand are both bound to the (trans)membrane of the cell it's cascade is activated through cell-cell interactions. These cell-cell interactions are disturbed due to the presence of cells with the mutant EFNB1 gene, as a result causing incomplete tissue-border formation.
Paradoxical to other X-linked conditions, with CFND the females are more severely affected than males. This is due to the process of X-inactivation in females, where at random either the maternal or paternal X-chromosome is inactivated in a cell. Due to this process the body’s tissues contain either cells with normal EFNB1 or the mutated EFNB1. This is called a mosaic pattern. This mosaic pattern of cells 'interferes' with the functionality of the cell-cell interactions, as a result causing the severe physical malformations in females.
As with all X-linked conditions CFND has a preset chance of being passed down from parents to their offspring. Females have two X-chromosomes and males have one X-chromosome. When a mother is a carrier of CFND, there is a 50% chance of her passing down the X-chromosome containing the mutated EFNB1 gene to her offspring, regardless if the child is a boy or girl. If the father is a carrier there is a 100% chance of him passing down his X-chromosome with the EFNB1 mutation to a daughter, and 0% chance of him passing it down to a son.
Diagnosis
The diagnosis CFND is established only after the presence of a mutation in the EFNB1 gene has been determined. Physical manifestations are not necessarily part of the diagnostic criteria, but can help guide in the right direction. This is due to the large heterogeneity between patients regarding phenotypic expression.
20% of the patients that present with CFND-like characteristics do not display a mutation in the EFNB1 gene. The group of patients diagnosed with CFND is thus often overestimated. However, it is important to distinguish this population from CFND for research purposes. On the other hand, especially in males, it is possible that someone is a carrier of the EFNB1 gene mutation yet does not present with any physical manifestations. Screening for the presence of an EFNB1 mutation is thus the most reliable method to establish the diagnosis CFND.
Genetic counseling or prenatal screening may be advised if there is a reason to suspect the presence of an EFNB1 gene mutation. Prenatal screening may be done by performing an ultrasound, where can be searched specifically for hypertelorism or a bifid nasal tip. However, this is quite difficult as facial involvement may not be obvious at such an early age, especially in cases with mild phenotypic presentation. The most definitive way to prove the presence of CFND is done by genetic testing, through amniocentesis and chorionic villus sampling. This however carries a greater risk of premature termination of the pregnancy.
Treatment
There is no ‘standard treatment’ for people with CFND due to the large variations in phenotypic expression. Each patient needs to be assessed and treated based on their specific presentation in order to restore the aesthetic and functional balance.
Surgical corrections for the main symptoms;